SAMENVATTING VAN DE PRODUCTKENMERKEN
![]() Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
1. NAAM VAN HET GENEESMIDDEL
Vabysmo 120 mg/ml oplossing voor injectie
Vabysmo 120 mg/ml oplossing voor injectie in voorgevulde spuit
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Eén ml oplossing voor injectie bevat 120 mg faricimab.
Voorgevulde spuit
Elke voorgevulde spuit bevat 21 mg faricimab in 0,175 ml oplossing. Dit levert een bruikbare hoeveelheid om een enkele dosis toe te dienen van 0,05 ml oplossing met 6 mg faricimab.
Injectieflacon
Elke injectieflacon bevat 28,8 mg faricimab in 0,24 ml oplossing. Dit levert een bruikbare hoeveelheid om een enkele dosis toe te dienen van 0,05 ml oplossing met 6 mg faricimab.
Faricimab is een gehumaniseerd antilichaam dat wordt geproduceerd in ovariumcellen van de Chinese hamster (CHO) door middel van DNA‑recombinatietechniek.
Hulpstoffen met bekend effect:
Elke 0,05 ml oplossing bevat 0,02 mg polysorbaat en 0,07 mg natrium.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie (injectie)
Heldere tot bijna doorschijnende, kleurloze tot bruinachtig gele oplossing, met een pH van 5,5 en een osmolaliteit van 270‑370 mOsm/kg.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Vabysmo is geïndiceerd voor de behandeling van volwassen patiënten met:
- neovasculaire (natte) leeftijdsgebonden maculadegeneratie (natte LMD)
- visusverslechtering als gevolg van diabetisch macula‑oedeem (DME);
- visusverslechtering als gevolg van macula-oedeem secundair aan retinale veneuze occlusies (RVO) (veneuze takocclusie (branch RVO) of retinale veneuze stamocclusie (central RVO)).
4.2 Dosering en wijze van toediening
Dit geneesmiddel mag alleen worden toegediend onder toezicht van een beroepsbeoefenaar in de gezondheidszorg die ervaring heeft met intravitreale injecties.
Dosering
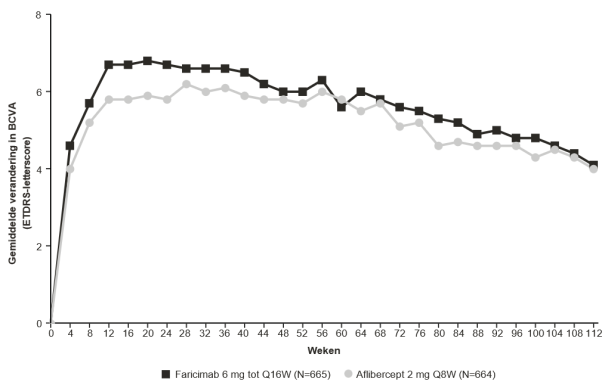
Neovasculaire (natte) leeftijdsgebonden maculadegeneratie (natte LMD)
De aanbevolen dosis is 6 mg (0,05 ml oplossing) toegediend als intravitreale injectie elke 4 weken (maandelijks) voor de eerste 3 doses.
Daarna wordt aanbevolen om 16 en/of 20 weken na de start van de behandeling de ziekteactiviteit op basis van anatomische en/of visuele resultaten te beoordelen, zodat de behandeling geïndividualiseerd kan worden. Bij patiënten zonder ziekteactiviteit moet toediening met faricimab elke 16 weken (4 maanden) worden overwogen. Bij patiënten met ziekteactiviteit moet behandeling elke 8 weken (2 maanden) of elke 12 weken (3 maanden) worden overwogen. Als anatomische en/of visuele resultaten veranderen, moet het behandelinterval hierop worden aangepast. Het interval moet worden verkort bij verslechtering van anatomische en/of visuele resultaten (zie rubriek 5.1). Er zijn beperkte gegevens beschikbaar over de veiligheid van behandelintervallen van 8 weken of korter tussen de injecties (zie rubriek 4.4). Controle tussen de bezoeken waarop toediening plaatsvindt moet worden gepland op basis van de status van de patiënt en beoordeling van de arts, maar er is geen vereiste voor maandelijkse controle tussen de injecties.
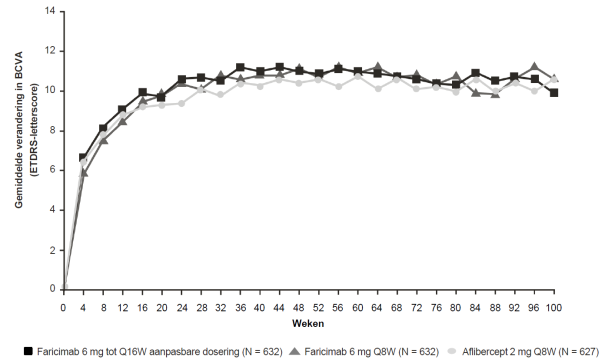
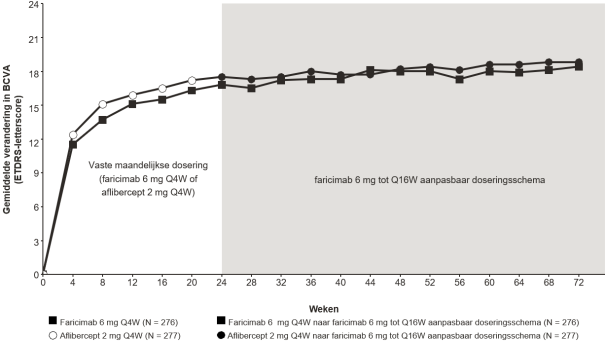
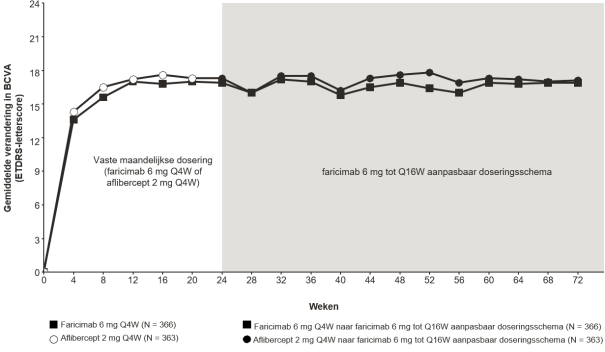
Visusverslechtering als gevolg van diabetisch macula-oedeem (DME) en macula-oedeem secundair aan retinale veneuze occlusie (RVO)
De aanbevolen dosis is 6 mg (0,05 ml oplossing) toegediend als intravitreale injectie elke 4 weken (maandelijks); 3 of meer opeenvolgende, maandelijkse injecties kunnen nodig zijn.
Daarna wordt de behandeling geïndividualiseerd volgens een treat-and-extend benadering. Op basis van de beoordeling van anatomische en/of visuele resultaten bij de patiënt door de arts, kan het behandelinterval worden verlengd in stappen van maximaal 4 weken. Als anatomische en/of visuele resultaten veranderen, moet het behandelinterval hierop worden aangepast. Het interval moet worden verkort bij verslechtering van anatomische en/of visuele resultaten (zie rubriek 5.1). Behandelintervallen korter dan 4 weken en langer dan 4 maanden tussen de injecties zijn niet onderzocht. Controle tussen de bezoeken waarop toediening plaatsvindt moet worden gepland op basis van de status van de patiënt en beoordeling van de arts, maar er is geen vereiste voor maandelijkse controle tussen de injecties.
Duur van de behandeling
Dit geneesmiddel is bedoeld voor langdurige behandeling. Als uit visuele en/of anatomische resultaten blijkt dat de patiënt geen baat heeft bij het voortzetten van de behandeling, moet de behandeling worden gestaakt.
Uitgestelde of gemiste dosis
Als een dosis wordt uitgesteld of gemist, moet de patiënt op het volgende ingeplande bezoek worden beoordeeld door de arts en afhankelijk van dit oordeel de dosering voortzetten.
Speciale populaties
Ouderen
Er is geen dosisaanpassing nodig bij patiënten van 65 jaar of ouder (zie rubriek 5.2). Er zijn beperkte gegevens over de veiligheid bij patiënten met natte LMD en RVO ≥ 85 jaar (zie rubriek 4.4).
Verminderde nierfunctie
Er is geen dosisaanpassing nodig bij patiënten met verminderde nierfunctie (zie rubriek 5.2).
Verminderde leverfunctie
Er is geen dosisaanpassing nodig bij patiënten met verminderde leverfunctie (zie rubriek 5.2).
Pediatrische patiënten
Er is geen relevante toepassing van Vabysmo bij pediatrische patiënten voor de indicaties natte LMD, DME en RVO.
Wijze van toediening
Uitsluitend voor intravitreaal gebruik. Elke voorgevulde spuit of injectieflacon mag uitsluitend worden gebruikt voor de behandeling van één oog.
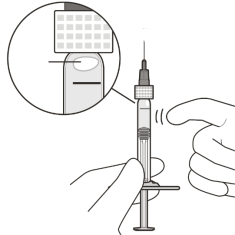
Vabysmo moet vóór toediening visueel worden geïnspecteerd op deeltjes en verkleuring en indien die aanwezig zijn, mag de voorgevulde spuit of injectieflacon niet worden gebruikt.
De intravitreale injectieprocedure moet onder aseptische omstandigheden worden uitgevoerd, waaronder chirurgische desinfectie van de handen, een steriel laken en een steriel ooglid‑speculum (of equivalent). De medische voorgeschiedenis van de patiënt voor overgevoeligheidsreacties moet zorgvuldig worden geëvalueerd voordat de intravitreale procedure wordt uitgevoerd (zie rubriek 4.8). Voorafgaand aan de injectie moet een geschikt anestheticum en een topisch breedspectrummicrobicide worden toegediend om de perioculaire huid, het ooglid en het oogoppervlak te desinfecteren.
Voorgevulde spuit
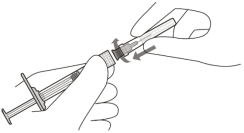
De voorgevulde spuit bevat een volume-overmaat. Het overtollige volume moet worden verwijderd vóór injectie van de aanbevolen dosis. Het injecteren van het gehele volume van de voorgevulde spuit kan leiden tot overdosering.
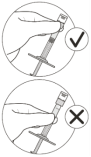
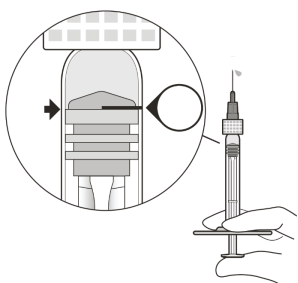
Om de luchtbellen samen met het overtollige geneesmiddel te verwijderen, drukt u de zuigerstang langzaam in totdat de onderrand van de ronde top van de rubberen zuigerstop zich op één lijn bevindt met de 0,05 ml dosismarkering (zie rubriek 4.9 en 6.6).
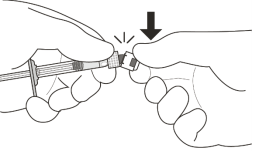
De injectienaald met filter (meegeleverd in de verpakking) moet worden ingebracht in de vitreale holte 3,5 tot 4,0 mm posterieur aan de limbus, waarbij de horizontale meridiaan moet worden vermeden en de naald moet worden gericht op het midden van de oogbol. Het injectievolume van 0,05 ml wordt dan langzaam toegediend; voor daaropvolgende injecties moet een andere positie op de sclera worden gebruikt.
Injectieflacon
De injectienaald (30-gauge x ½ inch, niet in de verpakking meegeleverd) moet worden ingebracht in de vitreale holte 3,5 tot 4,0 mm posterieur aan de limbus, waarbij de horizontale meridiaan moet worden vermeden en de naald moet worden gericht op het midden van de oogbol. Het injectievolume van 0,05 ml wordt dan langzaam toegediend; voor daaropvolgende injecties moet een andere positie op de sclera plaats worden gebruikt.
Controle na de injectie
Na de injectie moet al het ongebruikte geneesmiddel of afvalmateriaal worden vernietigd overeenkomstig lokale voorschriften.
Onmiddellijk na de intravitreale injectie moeten patiënten worden gecontroleerd op verhoging van de intraoculaire druk. Gepaste controle kan bestaan uit een controle op perfusie van de kop van de nervus opticus of tonometrie. Indien nodig moet steriele apparatuur voor paracentese beschikbaar zijn.
Na de intravitreale injectie moet patiënten worden geïnstrueerd om direct alle symptomen te melden wijzend op endoftalmitis (bijv. verlies van zicht, oogpijn, roodheid van het oog, fotofobie, wazig zien).
Voor instructies over het hanteren van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Actieve of vermoede oculaire of perioculaire infecties.
Actieve intraoculaire ontsteking.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest vaak gemelde bijwerkingen waren cataract (10%), conjunctivale bloeding (7%), glasvochtloslating (4%), toename in IOD (4%), mouches volantes (4%), oogpijn (3%) en retinale pigmentepitheelscheur (alleen bij natte LMD) (3%).
De meest ernstige bijwerkingen waren uveïtis (0,5%), endoftalmitis (0,4%), vitritis (0,4%), retinale scheur (0,2%), rhegmatogene netvliesloslating (0,1%) en traumatisch cataract (< 0,1%) (zie rubriek 4.4).
Tabel met lijst van bijwerkingen
De bijwerkingen die werden gemeld in klinische onderzoeken of na het op de markt brengen worden weergegeven volgens MedDRA-systeem/orgaanklasse en gerangschikt per frequentie waarbij de volgende conventie is gebruikt: Zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000) of niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiecategorie worden bijwerkingen weergegeven in volgorde van afnemende ernst.
Tabel 1: Frequenties van bijwerkingen
MedDRA‑systeem/orgaanklasse | Frequentiecategorie |
Oogaandoeningen | |
Cataract | Vaak |
Conjunctivale bloeding | Vaak |
Glasvochtloslating | Vaak |
Verhoogde intraoculaire druk | Vaak |
Mouches volantes (glasvochttroebelingen) | Vaak |
Retinale pigmentepitheelscheur (alleen natte LMD) | Vaak |
Oogpijn | Vaak |
Cornea geschaafd | Soms |
Oogirritatie | Soms |
Verhoogde lacrimatie | Soms |
Wazig zien | Soms |
Oogpruritus | Soms |
Oculair ongemak | Soms |
Oculaire hyperemie | Soms |
Iritis | Soms |
Afname gezichtsscherpte | Soms |
Uveïtis | Soms |
Endoftalmitis | Soms |
Gevoel van vreemd lichaam | Soms |
Glasvochtbloeding | Soms |
Vitritis | Soms |
Iridocyclitis | Soms |
Conjunctivale hyperemie | Soms |
Pijn door ingreep | Soms |
Retinale scheur | Soms |
Rhegmatogene netvliesloslating | Soms |
Tijdelijke afname gezichtsscherpte | Zelden |
Traumatisch cataract | Zelden |
Retina vasculitis* | Niet bekend |
Retinale occlusieve vasculitis* | Niet bekend |
Termen die gemarkeerd zijn met een asterisk (*) zijn bijwerkingen die zijn vastgesteld op basis van spontane meldingen na het op de markt brengen. Omdat deze bijwerkingen op vrijwillige basis worden gemeld door een populatie van een onbekende omvang, is het niet altijd mogelijk om een betrouwbare schatting te maken van de frequentie.
Beschrijving van geselecteerde bijwerkingen
Retina vasculitis en retinale occlusieve vasculitis
Zeldzame gevallen van retina vasculitis en/of retinale occlusieve vasculitis werden spontaan gemeld na het in de handel brengen (zie rubriek 4.4). Retina vasculitis en retinale occlusieve vasculitis zijn ook gemeld door patiënten die intravitreale behandelingen kregen.
Productklasse-gerelateerde bijwerkingen
Er is een theoretisch risico op arteriële trombo-embolische voorvallen, waaronder beroerte en myocardinfarct, na intravitreaal gebruik van VEGF-remmers. Een lage incidentie van arteriële trombo-embolische voorvallen werd waargenomen in de klinische onderzoeken met faricimab bij patiënten met natte LMD, DME en RVO (zie rubriek 4.4). Bij alle indicaties werd geen merkbaar verschil waargenomen tussen de armen die werden behandeld met faricimab en de comparator.
Immunogeniteit
Er bestaat een kans op een immuunrespons bij patiënten die worden behandeld met faricimab (zie rubriek 4.4). Na behandeling met faricimab tot 112 weken (natte LMD), tot 100 weken (DME) en tot 72 weken (RVO), werden tijdens de behandeling ontstane antilichamen tegen faricimab gedetecteerd bij ongeveer 13,8%, 9,6% en 14,4% van de patiënten met respectievelijk natte LMD, DME en RVO, die gerandomiseerd waren naar faricimab. De klinische relevantie van antilichamen tegen faricimab op de veiligheid is op dit moment onduidelijk. De incidentie van intraoculaire ontsteking bij patiënten met antilichamen tegen faricimab was 12/98 (12,2%; natte LMD), 15/128 (11,7%; DME) en 9/95 (9,5%; RVO) en bij patiënten zonder antilichamen tegen faricimab 8/562 (1,4%; natte LMD), 5/1.124 (0,4%; DME) en 10/543 (1,8%; RVO). De incidentie van ernstige oculaire bijwerkingen bij patiënten met antilichamen tegen faricimab was 6/98 (6,1%; natte LMD), 14/128 (10,9%; DME) en 7/95 (7,4%; RVO) en bij patiënten zonder antilichamen tegen faricimab 23/562 (4,1%; natte LMD), 45/1.124 (4,0% DME) en 34/543 (6,3%; RVO). Antilichamen tegen faricimab werden niet geassocieerd met een impact op de klinische werkzaamheid of systemische farmacokinetiek.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden (zie hieronder voor details).
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be

7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Roche Registratie GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/22/1683/001
EU/1/22/1683/002
10. DATUM VAN HERZIENING VAN DE TEKST
8 mei 2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4624631 | Vabysmo 120 mg/ml oplossing voor injectie | € 953,6 | - | Ja | - | - |