SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Ocrevus 300 mg concentraat voor oplossing voor infusie
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke injectieflacon bevat 300 mg ocrelizumab in 10 ml met een concentratie van 30 mg/ml. De uiteindelijke concentratie van het geneesmiddel na verdunning bedraagt ongeveer 1,2 mg/ml.
Ocrelizumab is een gehumaniseerd monoklonaal antilichaam, dat wordt geproduceerd in Chinese hamsterovariumcellen door middel van DNA-recombinatietechniek.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Concentraat voor oplossing voor infusie.
Heldere tot enigszins doorschijnende en kleurloze tot lichtbruine oplossing.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Ocrevus is geïndiceerd voor de behandeling van volwassen patiënten met actieve relapsing multiple sclerose (RMS) die is gedefinieerd door klinische of beeldvormende kenmerken (zie rubriek 5.1).
Ocrevus is geïndiceerd voor de behandeling van volwassen patiënten met vroege primair progressieve multiple sclerose (PPMS) gebaseerd op de ziekteduur, mate van invaliditeit en beeldvormende kenmerken die typerend zijn voor ontstekingsactiviteit (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
Behandeling moet gestart en gecontroleerd worden door gespecialiseerde artsen met ervaring in de diagnose en behandeling van neurologische aandoeningen en met toegang tot gepaste medische ondersteuning om ernstige reacties zoals ernstige infusiegerelateerde reacties (IRR’s) te kunnen behandelen.
Premedicatie voor infusiegerelateerde reacties
Voorafgaand aan elke infusie met ocrelizumab moeten de volgende twee soorten premedicatie toegediend worden om de frequentie en ernst van IRR’s te verminderen (zie in rubriek 4.4 voor aanvullende informatie om IRR’s te verminderen):
100 mg intraveneuze methylprednisolon (of een equivalent ervan), ongeveer 30 minuten voorafgaand aan elke infusie;
antihistaminicum ongeveer 30-60 minuten voorafgaand aan elke infusie.
Premedicatie met een antipyreticum (bijv. paracetamol), ongeveer 30-60 minuten voorafgaand aan elke infusie met Ocrevus kan eveneens worden overwogen.
Dosering:
Initiële dosis
De initiële dosis van 600 mg wordt toegediend in twee afzonderlijke intraveneuze infusies; eerst als een infusie van 300 mg, 2 weken later gevolgd door een tweede infusie van 300 mg (zie tabel 1).
Volgende doses
Alle daaropvolgende doses van ocrelizumab worden elke 6 maanden toegediend als één enkele intraveneuze infusie van 600 mg (zie tabel 1). De eerste dosis van 600 mg moet 6 maanden na de eerste infusie van de initiële dosis toegediend worden.
Er moet een periode van minstens 5 maanden aangehouden worden tussen elke dosis van ocrelizumab.
Aanpassingen van de infusie in het geval van IRR’s
Levensbedreigende IRR’s
Als er verschijnselen zijn van een levensbedreigende of invaliderende IRR tijdens een infusie, zoals acute overgevoeligheid of acute respiratory distress syndrome (ARDS), dan moet de infusie onmiddellijk worden stopgezet en moet de patiënt op gepaste wijze behandeld worden. Behandeling moet bij deze patiënten permanent stopgezet worden (zie rubriek 4.3).
Ernstige IRR’s
Als een patiënt een ernstige IRR (zoals dyspneu) ondervindt, of een combinatie van symptomen met blozen, koorts en keelpijn, dan moet de infusie onmiddellijk onderbroken worden en moeten deze symptomen behandeld worden. De infusie mag alleen opnieuw opgestart worden wanneer alle symptomen verdwenen zijn. De infusiesnelheid bij het opnieuw opstarten moet de helft bedragen van de infusiesnelheid op het moment waarop de reactie begon. Er is geen aanpassing van de infusie nodig voor volgende nieuwe infusies, tenzij de patiënt een IRR ondervindt.
Lichte tot matige IRR’s
Als een patiënt een lichte tot matige IRR (bijv. hoofdpijn) ondervindt, dan moet de infusiesnelheid verlaagd worden naar de helft van de snelheid op het moment waarop de reactie begon. Deze verlaagde snelheid moet minstens 30 minuten aangehouden worden. Indien goed verdragen, mag daarna de infusiesnelheid verhoogd worden naar de initiële infusiesnelheid. Er is geen aanpassing van de infusie nodig voor volgende nieuwe infusies, tenzij de patiënt een IRR ondervindt.
Dosisaanpassingen tijdens de behandeling
Bovenstaande voorbeelden van dosisonderbrekingen en verlaging van de snelheid (voor lichte tot matige IRR’s) leiden tot een wijziging in de infusiesnelheid en -duur, maar niet van de totale dosis.
Dosisverlagingen worden niet aangeraden.
Uitgestelde of gemiste doses
Als een infusie wordt gemist, dan moet deze zo snel mogelijk toegediend worden; wacht niet tot de volgende geplande dosis. Het behandelinterval van 6 maanden (met een minimum van 5 maanden) tussen de doses moet worden aangehouden (zie tabel 1).
Speciale populaties
Volwassenen ouder dan 55 jaar
Gebaseerd op de beperkte beschikbare gegevens (zie rubrieken 5.1 en 5.2) is een aanpassing van de dosis niet nodig voor patiënten ouder dan 55 jaar. Patiënten die aan klinische onderzoeken deelnamen kregen nadat zij ouder dan 55 jaar waren geworden nog steeds elke 6 maanden 600 mg ocrelizumab.
Verminderde nierfunctie
De veiligheid en werkzaamheid van ocrelizumab bij patiënten met een verminderde nierfunctie werd niet formeel onderzocht. Patiënten met een licht verminderde nierfunctie werden in klinische onderzoeken geïncludeerd. Er is geen ervaring met patiënten met matig tot ernstig verminderde nierfunctie. Ocrelizumab is een monoklonaal antilichaam dat wordt geklaard via katabolisme (d.w.z. afbraak in peptiden en aminozuren). Een dosisaanpassing is naar verwachting niet nodig voor patiënten met een verminderde nierfunctie (zie rubriek 5.2).
Verminderde leverfunctie
De veiligheid en werkzaamheid van ocrelizumab bij patiënten met een verminderde leverfunctie werd niet formeel onderzocht. Patiënten met een licht verminderde leverfunctie werden in klinische onderzoeken geïncludeerd. Er is geen ervaring met patiënten met matig tot ernstig verminderde leverfunctie. Ocrelizumab is een monoklonaal antilichaam dat wordt geklaard via katabolisme (in plaats van levermetabolisme). Een dosisaanpassing is naar verwachting niet nodig voor patiënten met verminderde leverfunctie (zie rubriek 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van ocrelizumab bij kinderen en adolescenten van 0 tot 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Ocrevus 300 mg concentraat voor oplossing voor infusie is niet bedoeld voor subcutaan gebruik en mag alleen via een intraveneuze infusie toegediend worden.
Het is van belang om de etikettering op het product te controleren om er zeker van te zijn dat de juiste formulering (intraveneus of subcutaan) aan de patiënt gegeven wordt via de correcte toedieningsweg, zoals voorgeschreven.
Patiënten kunnen de behandeling starten met de intraveneuze of subcutane formulering van ocrelizumab.
Na verdunning wordt het product toegediend als een intraveneuze infusie via een lijn uitsluitend bedoeld voor dit product. Infusies mogen niet toegediend worden als intraveneuze push of bolus.
Als een patiënt geen ernstige IRR heeft ondervonden bij een eerdere infusie met ocrelizumab, kan een kortere (2 uur) infusieduur gehanteerd worden bij volgende doses (zie tabel 1, optie 2).
Tabel 1: Dosis en schema van ocrelizumab
| Hoeveelheid toe te dienen ocrelizumab | Instructies voor infusie | |
Initiële dosis | Infusie 1 | 300 mg in 250 ml | Start de infusie met een snelheid van 30 ml/uur gedurende 30 minuten |
Infusie 2 | 300 mg in 250 ml | ||
Volgende doses | Optie 1 | 600 mg in 500 ml | Start de infusie met een snelheid van 40 ml/uur gedurende 30 minuten |
OF | |||
Optie 2 | 600 mg in 500 ml | Start de infusie met een snelheid van 100 ml/uur gedurende de eerste 15 minuten | |
Oplossingen voor intraveneuze infusie worden bereid door verdunning van het concentraat in een infusiezak met 9 mg/ml (0,9%) natriumchloride-oplossing voor infusie naar een uiteindelijke ocrelizumabconcentratie van ongeveer 1,2 mg/ml.
Voor instructies over verdunning van het geneesmiddel voorafgaand aan de toediening, zie rubriek 6.6.
Patiënten moeten tijdens de infusie en tot minstens één uur na het voltooien van de infusie geobserveerd worden (zie rubriek 4.4).
4.3 Contra-indicaties
- Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Patiënten die actieve infecties hebben (zie rubriek 4.4).
Patiënten die ernstig immuungecompromitteerd zijn (rubriek 4.4).
Patiënten met bekende actieve maligniteiten (zie rubriek 4.4).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
In de gecontroleerde periode van de klinische registratie-onderzoeken waren de belangrijkste en vaak gemelde bijwerkingen IRR’s (respectievelijk 34,3% en 40,1% in RMS en PPMS) en infecties (respectievelijk 58,5% en 72,2% in RMS en PPMS) (zie rubriek 4.4).
In totaal werden 2.376 patiënten geïncludeerd in de gecontroleerde periode van de klinische registratie-onderzoeken; 1.852 van deze patiënten gingen de OLE-fase in. Alle patiënten stapten over op behandeling met ocrelizumab tijdens de OLE-fase. De OLE-fase werd door 1.155 patiënten voltooid, wat resulteerde in een continue behandeling met ocrelizumab van ongeveer 10 jaar (blootstelling van 15.515 patiëntjaren) tijdens de gecontroleerde periode en de OLE-fase. Het totale veiligheidsprofiel dat tijdens de gecontroleerde periode en de OLE-fase werd waargenomen, blijft consistent met het veiligheidsprofiel dat werd waargenomen tijdens de gecontroleerde periode.
Lijst van bijwerkingen in tabelvorm
Bijwerkingen die gemeld zijn in de gecontroleerde periode van de klinische registratie-onderzoeken en bijwerkingen van spontane meldingen zijn opgesomd in tabel 2. De bijwerkingen zijn weergegeven per systeem/orgaanklasse en frequentiecategorie volgens MedDRA. Frequenties worden gedefinieerd als zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke systeem/orgaanklasse zijn de bijwerkingen vermeld op volgorde van afnemende frequentie.
Tabel 2 Bijwerkingen
| Zeer vaak | Vaak | Niet bekend2 |
Infecties en parasitaire aandoeningen | Infectie van de bovenste luchtwegen, nasofaryngitis, griep | Sinusitis, bronchitis, orale herpes, gastro-enteritis, luchtweginfectie, virale infectie, herpes zoster, conjunctivitis, cellulitis |
|
Bloed- en lymfestelselaandoeningen |
| Neutropenie | Laat optredende neutropenie2 |
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen |
| Hoesten, catarre |
|
Onderzoeken | Verlaagd immunoglobuline M in het bloed | Verlaagd immunoglobuline G in het bloed |
|
Letsels, intoxicaties en verrichtingscomplicaties | Infusiegerelateerde reacties1 |
|
|
1 Zie ‘Beschrijving van geselecteerde bijwerkingen’.
2 Waargenomen na het op de markt brengen.
Beschrijving van geselecteerde bijwerkingen
Infusiegerelateerde reacties
In alle onderzoeken bij RMS- en PPMS-patiënten kwamen onder meer, maar niet uitsluitend, de volgende symptomen voor die geassocieerd worden met IRR’s: pruritus, huiduitslag, urticaria, erytheem, blozen, hypotensie, pyrexie, vermoeidheid, hoofdpijn, duizeligheid, geïrriteerde keel, orofaryngeale pijn, dyspneu, faryngeaal of laryngeaal oedeem, misselijkheid, tachycardie. In de gecontroleerde onderzoeken waren er geen fatale IRR’s. Na het op de markt brengen is ook anafylaxie gemeld als symptoom van IRR’s.
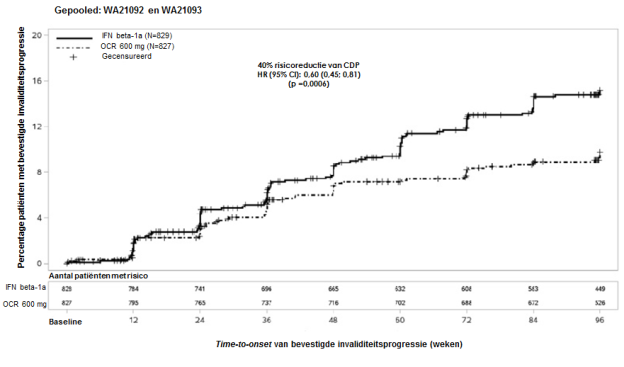
In de actief gecontroleerde (RMS) klinische onderzoeken was IRR de meest voorkomende bijwerking bij patiënten die behandeld werden met ocrelizumab met een totale incidentie van 34,3% vergeleken met een incidentie van 9,9% in de groep die met interferon bèta-1a behandeld werd (placebo-infusie). De incidentie van IRR’s was het hoogst tijdens dosis 1, infusie 1 (27,5%) en daalde gedurende de tijd tot < 10% bij dosis 4. De meerderheid van IRR’s in beide behandelgroepen waren licht tot matig in ernst. Bij patiënten die met ocrelizumab behandeld werden ondervond 21,7% van de patiënten lichte IRR’s en 10,1% van de patiënten matige IRR’s, 2,4% ondervond ernstige IRR’s en 0,1% ondervond levensbedreigende IRR’s.
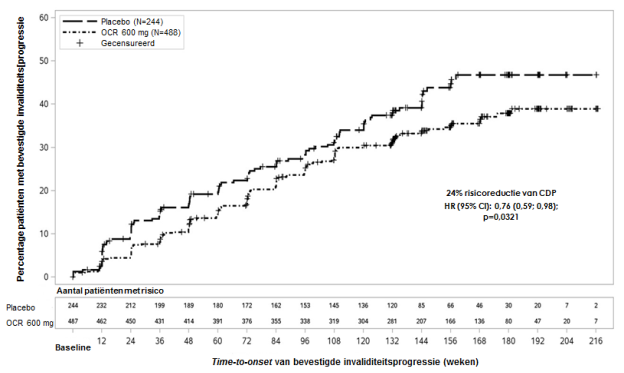
In het placebogecontroleerde (PPMS) klinische onderzoek, was IRR de meest voorkomende bijwerking bij patiënten die met ocrelizumab werden behandeld, met een totale incidentie van 40,1% vergeleken met een incidentie van 25,5% in de placebogroep. De incidentie van IRR’s was het hoogst tijdens dosis 1, infusie 1 (27,4%) en daalde bij volgende doses tot < 10% bij dosis 4. Een groter percentage patiënten in elke groep ondervond IRR’s tijdens de eerste infusie van elke dosis in vergelijking met de tweede infusie van die dosis. De meerderheid van IRR’s waren licht tot matig in ernst. Bij patiënten die met ocrelizumab behandeld werden, ondervond 26,7% van de patiënten lichte IRR’s en 11,9% van de patiënten matige IRR’s, 1,4% ondervond ernstige IRR’s. Er waren geen levensbedreigende IRR’s. Zie rubriek 4.4.
Gedurende de gecontroleerde periode en de OLE-fase van de klinische onderzoeken bij RMS- en PPMS-patiënten, kregen de patiënten ongeveer 20 doses ocrelizumab. De incidentie van IRR’s bij RMS-patiënten daalde tot < 4% tijdens dosis 4 van de OLE-fase en tot < 5% bij PPMS-patiënten tijdens dosis 5 van de OLE-fase. Bij volgende doses toegediend tijdens de OLE-fase bleef de incidentie van IRR laag. De meerderheid van IRR’s waren licht tijdens de OLE-fase.
Alternatieve kortere infusieduur bij vervolgdoses
In een onderzoek (MA30143 deelonderzoek naar kortere infusieduur) dat opgezet werd om het veiligheidsprofiel van kortere (2 uur durende) infusies met ocrelizumab bij patiënten met actieve relapsing-remitting multiple sclerose vast te stellen, kwamen de incidentie, intensiteit en type van de IRR-symptomen overeen met die bij 3,5 uur durende infusies (zie rubriek 5.1). Over het algemeen was het aantal interventies dat nodig was laag in beide infusiegroepen. Er waren echter meer interventies (verlagen van de snelheid of tijdelijk onderbreken) nodig om de IRR’s onder controle te krijgen in de groep met kortere (2 uur) infusieduur vergeleken met de groep die 3,5 uur durende infusies kregen (respectievelijk 8,7% versus 4,8%).
Infectie
In de actief gecontroleerde onderzoeken bij RMS-patiënten traden infecties op bij 58,5% van de patiënten die ocrelizumab kregen versus 52,5% van de patiënten die interferon bèta-1a kregen. Ernstige infecties traden op bij 1,3% van de patiënten die ocrelizumab kregen versus 2,9% van de patiënten die interferon bèta-1a kregen. In het placebogecontroleerde onderzoek bij PPMS-patiënten traden infecties op bij 72,2% van de patiënten die ocrelizumab kregen versus 69,9% van de patiënten die placebo kregen. Ernstige infecties traden op bij 6,2% van de patiënten die ocrelizumab kregen versus 6,7% van de patiënten die placebo kregen.
Alle patiënten stapten over op ocrelizumab gedurende de OLE-fase van de RMS- en PPMS-onderzoeken. Tijdens de OLE-fase nam het algehele risico op ernstige infecties bij RMS- en PPMS-patiënten niet toe ten opzichte van wat werd waargenomen tijdens de gecontroleerde periode. Zoals waargenomen tijdens de gecontroleerde periode, bleef de incidentie van ernstige infecties bij PPMS-patiënten hoger dan werd waargenomen bij RMS-patiënten.
In lijn met de eerdere analyse van risicofactoren voor ernstige infecties bij andere auto-immuunaandoeningen dan MS (zie rubriek 4.4), werd een multivariate analyse van risicofactoren voor ernstige infecties uitgevoerd in de ongeveer 10 jaar van cumulatieve blootstellingsgegevens uit de gecontroleerde periode en OLE-fase van de klinische registratie-onderzoeken. Risicofactoren voor ernstige infecties bij RMS‑patiënten zijn ten minste 1 comorbiditeit, recent klinisch recidief en Expanded Disability Status Scale (EDSS) ≥ 6,0. Risicofactoren voor ernstige infecties bij PPMS‑patiënten zijn onder andere een body mass index hoger dan 25 kg/m2, het hebben van ten minste 2 comorbiditeiten, EDSS ≥ 6,0 en IgM < lower limit of normal (LLN). Comorbiditeiten waren onder andere, maar niet uitsluitend, cardiovasculaire aandoeningen, nier- en urinewegaandoeningen, eerdere infecties en depressie.
Luchtweginfecties
Het percentage luchtweginfecties was hoger bij patiënten die met ocrelizumab werden behandeld in vergelijking met interferon bèta-1a en placebo.
In de klinische onderzoeken bij RMS-patiënten ondervond 39,9% van de patiënten die met ocrelizumab werden behandeld en 33,2% van de patiënten die met interferon bèta-1a werden behandeld een infectie van de bovenste luchtwegen en 7,5% van de patiënten die met ocrelizumab werden behandeld en 5,2% van de patiënten die met interferon bèta-1a werden behandeld ondervond een infectie van de onderste luchtwegen.
In het klinisch onderzoek bij PPMS-patiënten ondervond 48,8% van de patiënten die met ocrelizumab werden behandeld en 42,7% van de patiënten die placebo kregen een infectie van de bovenste luchtwegen, en 9,9% van de patiënten die met ocrelizumab werden behandeld en 9,2% van de patiënten die placebo kregen ondervond een infectie van de onderste luchtwegen.
De luchtweginfecties die werden gemeld bij patiënten die met ocrelizumab werden behandeld waren voornamelijk licht tot matig (80 - 90%).
Herpes
In actief gecontroleerde (RMS) klinische onderzoeken, werden herpesinfecties vaker gemeld bij patiënten die met ocrelizumab werden behandeld dan bij patiënten die met interferon bèta-1a werden behandeld, bijvoorbeeld herpes zoster (2,1% versus 1,0%), herpes simplex (0,7% versus 0,1%), orale herpes (3,0% versus 2,2%), genitale herpes (0,1% versus 0%) en herpesvirusinfectie (0,1% versus 0%). Alle Infecties waren licht tot matig in ernst, uitgezonderd van een geval met graad 3, en patiënten herstelden onder behandeling met standaardbehandelingen.
In het placebogecontroleerde (PPMS) klinische onderzoek werd een hoger percentage patiënten met orale herpes (2,7% versus 0,8%) waargenomen in de behandelarm die met ocrelizumab werd behandeld.
Afwijkende laboratoriumwaarden
Immunoglobulines
Behandeling met ocrelizumab leidde tot een daling in totaal immunoglobulines gedurende de gecontroleerde periode van de klinische registratie-onderzoeken, voornamelijk gedreven door een daling in IgM.
Gegevens uit de gecontroleerde periode en de OLE-fase van de klinische registratie-onderzoeken hebben een verband aangetoond tussen verlaagde IgG-spiegels (en in mindere mate voor IgM of IgA) en een verhoogde incidentie van ernstige infecties. Van de RMS-patiënten had 2,1% een ernstige infectie gedurende een periode met IgG < LLN, en had 2,3% van de PPMS-patiënten een ernstige infectie gedurende een periode met IgG < LLN. Het verschil in incidentie van ernstige infecties tussen patiënten met IgG < LLN en patiënten met IgG ≥ LLN nam in de loop van de tijd niet toe. Het type, de ernst, de latentie, de duur en de uitkomst van ernstige infecties die werden waargenomen tijdens episoden van immunoglobulinen onder LLN, kwamen overeen met de totale ernstige infecties die werden waargenomen bij patiënten die met ocrelizumab werden behandeld gedurende de gecontroleerde periode en de OLE-fase. Gedurende de 10 jaar van continue behandeling met ocrelizumab bleven de gemiddelde IgG-spiegels bij RMS- en PPMS-patiënten boven de LLN.
Lymfocyten
Bij RMS werd een daling in de lymfocytenwaarden < LLN waargenomen bij 20,7% van de patiënten die met ocrelizumab werden behandeld, in vergelijking met 32,6% van de patiënten die met interferon bèta-1a werden behandeld. Bij PPMS-patiënten werd een daling van lymfocytenwaarden < LLN waargenomen bij 26,3% van de patiënten die met ocrelizumab werden behandeld, in vergelijking met 11,7% van de patiënten die placebo kregen.
De meerderheid van de dalingen die bij patiënten die met ocrelizumab werden behandeld waren gemeld hadden een ernst van graad 1 (< LLN tot 800 cellen/mm3) en graad 2 (tussen 500 en 800 cellen/mm3). Ongeveer 1% van de patiënten in de groep die ocrelizumab kreeg, had lymfopenie van graad 3 (tussen 200 en 500 cellen/mm3). Geen van de patiënten had lymfopenie van graad 4 (< 200 cellen/mm3).
Een verhoogde incidentie van ernstige infecties werd waargenomen tijdens episoden van bevestigde verlaagde lymfocytenwaarden bij patiënten die met ocrelizumab werden behandeld. Het aantal ernstige infecties was te laag om definitieve conclusies te trekken.
Neutrofielen
In de actief gecontroleerde (RMS) behandelperiode werd een daling in de neutrofielenwaarden < LLN waargenomen bij 14,7% van de patiënten die met ocrelizumab werden behandeld in vergelijking met 40,9% van de patiënten die met interferon bèta-1a werden behandeld. In het placebogecontroleerde (PPMS) klinische onderzoek was het percentage patiënten dat een daling in de neutrofielenwaarden vertoonde hoger (12,9%) bij patiënten die met ocrelizumab werden behandeld dan bij patiënten die placebo kregen (10,0%). Van deze patiënten had een hoger percentage (4,3%) van de patiënten die met ocrelizumab werden behandeld een neutropenie van graad 2 of hoger ten opzichte van 1,3% in de groep die placebo kreeg. Ongeveer 1% van de patiënten die met ocrelizumab werd behandeld had een graad 4 neutropenie versus 0% in de groep die placebo kreeg.
De meerderheid van de dalingen in neutrofielen waren tijdelijk (slechts één keer waargenomen bij één bepaalde patiënt die met ocrelizumab werd behandeld) en hadden een ernst van graad 1 (tussen <LNN en < 1.500 cellen/mm3) en graad 2 (tussen 1.000 en 1.500 cellen/mm3). Over het geheel had ongeveer 1% van de patiënten in de groep die met ocrelizumab werd behandeld graad 3 of 4 neutropenie. Eén patiënt met neutropenie van graad 3 (tussen 500 en 1.000 cellen/mm3) en één patiënt met neutropenie van graad 4 (< 500 cellen/mm3) hadden een specifieke behandeling nodig met granulocyten-kolonie-stimulerende factor (G-CSF) en bleven onder behandeling met ocrelizumab na de episode. Neutropenie kan zich enkele maanden na toediening van ocrelizumab voordoen (zie rubriek 4.4).
Overig
Eén patiënt die 2.000 mg ocrelizumab had gekregen overleed na een MRI-onderzoek, 12 weken na de laatste infusie, aan systemisch inflammatoir responssyndroom (SIRS) met een onbekende oorzaak. Een anafylactische reactie op de MRI-gadoliniumcontrastvloeistof zou bijgedragen kunnen hebben aan het SIRS.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden (zie hieronder voor details).
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/17/1231/001
EU/1/17/1231/002
10. DATUM VAN HERZIENING VAN DE TEKST
13 februari 2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3519014 | OCREVUS 300MG CONC OPL INF 30MG/ML FL INJ 1X10ML | L04AA36 | - | € 4470,85 | Ja | - | - |