RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l'objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l'objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Columvi 2,5 mg solution à diluer pour perfusion
Columvi 10 mg solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Columvi 2,5 mg solution à diluer pour perfusion
Chaque flacon de 2,5 mL de solution à diluer contient 2,5 mg de glofitamab à une concentration de 1 mg/mL.
Columvi 10 mg solution à diluer pour perfusion
Chaque flacon de 10 mL de solution à diluer contient 10 mg de glofitamab à une concentration de 1 mg/mL.
Le glofitamab est un anticorps monoclonal bispécifique anti‑CD20/ anti‑CD3 humanisé produit dans des cellules d’ovaire de hamster chinois (CHO) par la technologie de l’ADN recombinant.
Excipients à effet notoire :
Chaque flacon de 2,5 mL de Columvi contient 1,25 mg (0,5 mg/mL) de polysorbate 20.
Chaque flacon de 10 mL de Columvi contient 5 mg (0,5 mg/mL) de polysorbate 20.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution à diluer pour perfusion (concentré stérile).
Solution limpide, incolore, de pH 5,5 et d’osmolalité 270-350 mOsm/kg.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
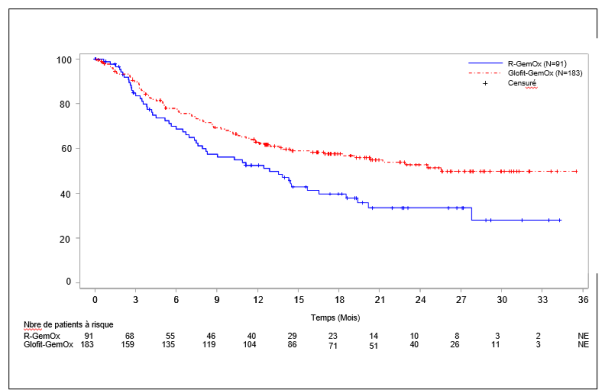
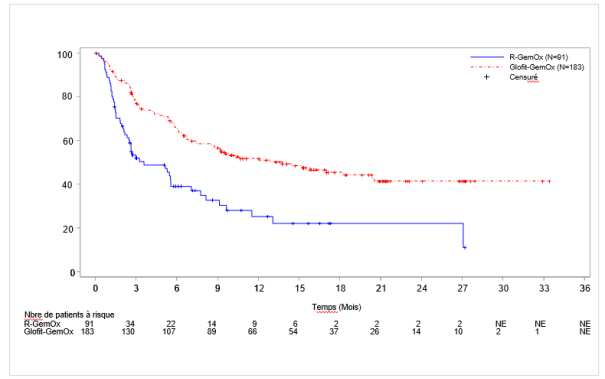
Columvi en association avec la gemcitabine et l’oxaliplatine est indiqué pour le traitement des patients adultes atteints d’un lymphome diffus à grandes cellules B non spécifié (LDGCB NOS) réfractaire ou en rechute, non éligibles à une autogreffe de cellules souches (ASCT).
Columvi en monothérapie est indiqué pour le traitement des patients adultes atteints d’un lymphome diffus à grandes cellules B (LDGCB) réfractaire ou en rechute, après au moins deux lignes de traitement systémique.
4.2 Posologie et mode d’administration
Columvi doit être administré exclusivement sous la supervision d'un professionnel de santé expérimenté dans le diagnostic et le traitement des patients atteints de cancer et ayant accès à une assistance médicale appropriée pour gérer les réactions sévères associées au syndrome de relargage des cytokines (SRC) et au syndrome de neurotoxicité associé aux cellules effectrices immunitaires (ICANS).
Au moins 1 dose de tocilizumab doit être disponible avant la perfusion de Columvi aux Cycles 1 et 2, cette dose pourrait être utilisée en cas de SRC. L’accès à une dose supplémentaire de tocilizumab dans les 8 heures suivant la précédente dose de tocilizumab doit être garanti (voir rubrique 4.4).
Prétraitement par obinutuzumab
Tous les patients de l’étude NP30179 et de l’étude GO41944 (STARGLO) ont reçu une dose unique de 1 000 mg d’obinutuzumab en prétraitement au Jour 1 du Cycle 1 (7 jours avant le début du traitement par Columvi) afin de diminuer le nombre de cellules B circulantes et lymphoïdes (voir Tableau 2, Doses retardées ou manquées, et rubrique 5.1).
L’obinutuzumab a été administré en perfusion intraveineuse à 50 mg/h. La vitesse de perfusion a été augmentée par paliers de 50 mg/h toutes les 30 minutes, jusqu’à un maximum de 400 mg/h.
Se reporter aux informations complètes de prescription concernant l’obinutuzumab pour en savoir plus sur la prémédication, la préparation, l’administration et la prise en charge des effets indésirables de l’obinutuzumab.
Prémédication et prophylaxie
Prophylaxie du syndrome de relargage des cytokines
Columvi doit être administré à des patients bien hydratés. La prémédication recommandée pour le SRC (voir rubrique 4.4) est décrite dans le Tableau 1.
Tableau 1. Prémédication avant la perfusion de Columvi
Cycle de traitement (Jour) | Patients nécessitant une prémédication | Prémédication | Administration |
Cycle 1 (Jour 8, Jour 15) ; | Tous les patients | Dexaméthasone 20 mg par voie intraveineuse1 | Terminée au moins 1 heure avant la perfusion de Columvi |
Antalgique / antipyrétique oral2 | Au moins 30 minutes avant la perfusion de Columvi | ||
Antihistaminique3 | |||
Toutes les perfusions ultérieures | Tous les patients | Antalgique / antipyrétique oral2 | Au moins 30 minutes avant la perfusion de Columvi |
Antihistaminique3 | |||
Patients ayant présenté un SRC avec la dose précédente | Dexaméthasone 20 mg par voie intraveineuse1, 4 | Terminée au moins 1 heure avant la perfusion de Columvi |
1 En cas d’intolérance à la dexaméthasone ou si la dexaméthasone n’est pas disponible, administrer 100 mg de prednisone/prednisolone ou 80 mg de méthylprednisolone.
2 Par exemple, 1 000 mg de paracétamol.
3 Par exemple, 50 mg de diphenhydramine.
4 A administrer en complément de la prémédication requise pour tous les patients.
Posologie
L’administration de Columvi commence par un schéma d’escalade de dose (destiné à réduire le risque de SRC) jusqu’à atteindre la dose recommandée de 30 mg.
Schéma d’escalade de dose de Columvi en monothérapie
Columvi doit être administré en perfusion intraveineuse conformément au schéma d’escalade de dose jusqu’à atteindre la dose recommandée de 30 mg (comme indiqué dans le Tableau 2), une fois que le prétraitement par obinutuzumab au Jour 1 du Cycle 1 est terminé. Chaque cycle dure 21 jours.
Tableau 2. Schéma d’escalade de dose de Columvi en monothérapie pour les patients atteints d’un LDGCB réfractaire ou en rechute
Cycle de traitement, Jour | Dose de Columvi | Durée de la perfusion | |
Cycle 1 | Jour 1 | Prétraitement par obinutuzumab 1 000 mg1 | |
Jour 8 | 2,5 mg | 4 heures2 | |
Jour 15 | 10 mg | ||
Cycle 2 | Jour 1 | 30 mg | |
Cycle 3 à 12 | Jour 1 | 30 mg | 2 heures3 |
1 Se reporter à « Prétraitement par obinutuzumab » ci-dessus. | |||
Schéma d’escalade de dose de Columvi en association avec la gemcitabine et l’oxaliplatine
Columvi doit être administré en perfusion intraveineuse conformément au schéma d’escalade de dose jusqu’à atteindre la dose recommandée de 30 mg (comme indiqué dans le Tableau 3), une fois que le prétraitement par obinutuzumab au Jour 1 du Cycle 1 est terminé.
Columvi est administré en association avec la gemcitabine et l’oxaliplatine aux Cycles 1 à 8 et en monothérapie aux Cycles 9 à 12. Chaque cycle dure 21 jours.
Tableau 3. Schéma d’escalade de dose de Columvi en association avec la gemcitabine et l’oxaliplatine pour les patients atteints d’un LDGCB réfractaire ou en rechute
Cycle de traitement, Jour | Dose de Columvi (durée de la perfusion) | Dose de gemcitabine | Dose d’oxaliplatine | |
Cycle 1 | Jour 1 | Prétraitement par obinutuzumab 1 000 mga | ||
Jour 2 | – | 1 000 mg/m2 b | 100 mg/m2 b | |
Jour 8 | 2,5 mg (4 heures)c | – | – | |
Jour 15 | 10 mg (4 heures)c | |||
Cycle 2 | Jour 1 | 30 mg (4 heures)c,d | 1 000 mg/m2 b, d | 100 mg/m2 b, d |
Cycle 3 à 8 | Jour 1 | 30 mg (2 heures)d,e | 1 000 mg/m2 b, d | 100 mg/m2 b, d |
Cycle 9 à 12 | Jour 1 | 30 mg (2 heures)e | – | – |
a Se reporter à « Prétraitement par obinutuzumab » ci-dessus.
b Cycle 1 à 8 : Administrer la gemcitabine avant l’oxaliplatine.
c Pour les patients présentant un SRC lors d’une administration précédente de Columvi, la durée de la perfusion peut être étendue jusqu’à 8 heures (voir rubrique 4.4).
d Cycles 2 à 8 : Administrer Columvi avant la gemcitabine et l’oxaliplatine. La gemcitabine et l’oxaliplatine peuvent être administrées le Jour 1 ou 2.
e La durée de la perfusion peut être raccourcie à 2 heures à la discrétion du médecin, si la perfusion précédente a été bien tolérée. Si le patient a présenté un SRC lors d’une administration précédente, la durée de la perfusion doit être maintenue à 4 heures.
Surveillance des patients
Lorsque Columvi est administré en monothérapie, les signes et symptômes d’un potentiel SRC doivent être surveillés chez les patients pendant la durée de toutes les perfusions de Columvi et pendant au moins 10 heures après la fin de la perfusion de la première dose de Columvi (2,5 mg au Jour 8 du Cycle 1) (voir rubrique 4.8).
Lorsque Columvi est administré en association avec la gemcitabine et l’oxaliplatine, les signes et symptômes d’un potentiel SRC doivent être surveillés chez les patients pendant la durée de toutes les perfusions de Columvi et pendant 4 heures après la fin de la perfusion de la première dose de Columvi (2,5 mg au Jour 8 du Cycle 1) (voir rubrique 4.8).
Les patients ayant présenté un SRC de Grade ≥ 2 lors de leur précédente perfusion doivent faire l’objet d’une surveillance après la fin de la perfusion (voir le Tableau 4 dans la rubrique 4.2).
Tous les patients doivent être surveillés afin de détecter les signes et symptômes du SRC et du syndrome de neurotoxicité associé aux cellules effectrices immunitaires (ICANS) après administration de Columvi.
Tous les patients doivent être informés du risque, des signes et des symptômes du SRC et de l’ICANS. Il doit leur être conseillé de contacter un professionnel de santé immédiatement en cas d'apparition de signes et symptômes de SRC et/ou d’ICANS à tout moment (voir rubrique 4.4).
Durée du traitement
La durée recommandée de traitement par Columvi en monothérapie est de 12 cycles au maximum, ou jusqu’à progression de la maladie ou apparition d’une toxicité inacceptable, selon la première éventualité. Chaque cycle dure 21 jours.
La durée recommandée de traitement par Columvi en association avec la gemcitabine et l’oxaliplatine est de 8 cycles, suivis de 4 cycles de Columvi en monothérapie pour 12 cycles au maximum de Columvi, ou jusqu’à progression de la maladie ou apparition d’une toxicité inacceptable, selon la première éventualité. Chaque cycle dure 21 jours.
Doses retardées ou manquées
Pendant le schéma d’escalade de dose (administration hebdomadaire) :
Après le prétraitement par obinutuzumab, si la dose de 2,5 mg de Columvi est retardée de plus d’1 semaine, le prétraitement par obinutuzumab doit être répété.
Après la dose de 2,5 mg ou de 10 mg de Columvi, en cas d’intervalle entre 2 doses de Columvi compris entre 2 et 6 semaines, répéter la dernière dose de Columvi bien tolérée et reprendre le schéma d’escalade de dose prévu.
Après la dose de 2,5 mg ou de 10 mg de Columvi, en cas d’intervalle entre 2 doses de Columvi de plus de 6 semaines, répéter le prétraitement par obinutuzumab et le schéma d’escalade de dose de Columvi (voir Cycle 1 dans le Tableau 2 et le Tableau 3).
Après le Cycle 2 (dose de 30 mg) :
En cas d’intervalle entre 2 cycles de Columvi de plus de 6 semaines, répéter le prétraitement par obinutuzumab et le schéma d’escalade de dose de Columvi (voir Cycle 1 dans le Tableau 2 et le Tableau 3), puis reprendre le cycle de traitement prévu (dose de 30 mg).
Modifications de la dose
Aucune réduction de dose de Columvi n’est recommandée.
Prise en charge du syndrome de relargage des cytokines
Le syndrome de relargage des cytokines doit être identifié en se basant sur le tableau clinique (voir rubriques 4.4 et 4.8). Les autres causes de fièvre, d’hypoxie et d’hypotension artérielle, comme une infection ou un sepsis, doivent être recherchées. Si un SRC est suspecté, il doit être pris en charge conformément aux recommandations de prise en charge du SRC d’après la classification de l’ASTCT (American Society for Transplantation and Cellular Therapy : Société américaine de transplantation et de thérapie cellulaire) indiquées dans le Tableau 4.
Tableau 4. Classification et recommandations de prise en charge du SRC selon l’ASTCT
Grade1 | Prise en charge du SRC | Pour la perfusion suivante prévue de Columvi |
Grade 1 | Si un SRC se produit pendant la perfusion : | S’assurer de la résolution des symptômes pendant au moins 72 heures avant la perfusion suivante |
Grade 2 | Si un SRC se produit pendant la perfusion : | S’assurer de la résolution des symptômes pendant au moins 72 heures avant la perfusion suivante |
Pour le Grade 2 : Utilisation de tocilizumab | ||
Grade 3 | Si un SRC se produit pendant la perfusion : | S’assurer de la résolution des symptômes pendant au moins 72 heures avant la perfusion suivante |
Grade 4 | Si un SRC se produit pendant ou après la perfusion : | |
Pour le Grade 3 et le Grade 4 : Utilisation de tocilizumab | ||
1 Classification de l’ASTCT (American Society for Transplantation and Cellular Therapy) (Lee 2019). | ||
Prise en charge du syndrome de neurotoxicité associé aux cellules effectrices immunitaires (ICANS)
Dès le premier signe d’ICANS et en fonction du type et de la gravité, envisager un traitement symptomatique, une évaluation neurologique et l’interruption du traitement par Columvi (voir Tableau 5). Écarter d’autres causes de symptômes neurologiques. En cas de suspicion d’ICANS, la prise en charge doit être conforme aux recommandations du Tableau 5.
Tableau 5. Classification et recommandations de prise en charge de l’ICANS
Grade1 | Symptômes présentés2 | Prise en charge de l’ICANS | |
SRC concomitant | Absence de SRC concomitant | ||
Grade 1 | Score ICE3 7-9 | Mettre en œuvre la prise en charge du SRC selon le Tableau 4. | Surveiller les symptômes neurologiques et envisager une consultation et une évaluation en neurologie, à la discrétion du médecin. |
Suspendre le traitement par Columvi jusqu’à résolution de l’ICANS. | |||
Grade 2 | Score ICE3 3-6 | Administrer du tocilizumab conformément au Tableau 4 relatif à la prise en charge du SRC. | Administrer 10 mg de dexaméthasone5 par voie intraveineuse toutes les 6 heures. |
Suspendre le traitement par Columvi jusqu’à résolution de l’ICANS. | |||
Grade 3 | Score ICE3 0-2 | Administrer du tocilizumab conformément au Tableau 4 relatif à la prise en charge du SRC. | Administrer 10 mg de dexaméthasone5 par voie intraveineuse toutes les 6 heures. |
Suspendre le traitement par Columvi jusqu’à résolution de l’ICANS. | |||
Grade 4 | Score ICE3 0 | Administrer du tocilizumab conformément au Tableau 4 relatif à la prise en charge du SRC. | Administrer 10 mg de dexaméthasone5 par voie intraveineuse toutes les 6 heures. |
Arrêtez définitivement Columvi. | |||
1 Critères de gradation consensuelle des ICANS de l’ASTCT (Lee 2019).
2 La prise en charge est déterminée par l’événement le plus grave, non imputable à toute autre cause.
3 Si le patient est éveillé et capable de faire l’objet d’une évaluation de l’encéphalopathie associée aux cellules effectrices immunitaires (ICE), évaluer les points suivants :
Orientation (le patient peut citer l’année, le mois, la ville, l’hôpital = 4 points) ;
Désignation (le patient doit donner le nom de 3 objets, le médecin désigne p. ex. une horloge, un stylo, un bouton = 3 points) ;
Réalisation de consignes (p. ex., «montrez-moi deux doigts» ou «fermez les yeux et tirez la langue» = 1 point) ;
Écriture (aptitude à écrire une phrase standard = 1 point) ;
Attention (compter à rebours et par 10 à partir de 100 = 1 point).
Si le patient n’est pas éveillé et ne peut faire l’objet d’aucune évaluation ICE (ICANS de grade 4) = 0 point.
4 Non imputable à toute autre cause.
5 Toutes les mentions d’administration de dexaméthasone font référence à de la dexaméthasone ou à des équivalents.
Populations particulières
Population âgée
Aucun ajustement posologique n’est requis chez les patients âgés de 65 ans et plus (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement posologique n’est requis chez les patients présentant une insuffisance hépatique légère (bilirubine totale supérieure à la limite supérieure de la normale [LSN] et ≤ 1,5 x LSN ou aspartate transaminase [ASAT] > LSN). Columvi n’a pas été étudié chez des patients présentant une insuffisance hépatique modérée ou sévère (voir rubrique 5.2).
Insuffisance rénale
Aucun ajustement posologique n’est requis chez les patients présentant une insuffisance rénale légère ou modérée (CLCr ≥ 30 et < 90 mL/min). Columvi n’a pas été étudié chez des patients présentant une insuffisance rénale sévère (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de Columvi chez les enfants âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Columvi est administré uniquement par voie intraveineuse.
Columvi doit être dilué par un professionnel de santé en utilisant une technique aseptique, avant administration intraveineuse. Il doit être administré en perfusion intraveineuse via une ligne de perfusion dédiée.
Columvi ne doit pas être administré en injection rapide ou bolus intraveineux.
Pour les instructions concernant la dilution de Columvi avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active, à l’obinutuzumab, ou à l’un des excipients mentionnés à la rubrique 6.1.
Pour les contre-indications spécifiques de l’obinutuzumab, se reporter aux informations de prescription de l’obinutuzumab.
4.8 Effets indésirables
Résumé du profil de sécurité
Columvi en monothérapie
Les effets indésirables les plus fréquents (≥ 20 %) ont été le syndrome de relargage des cytokines, la neutropénie, l’anémie, la thrombopénie et le rash.
Les effets indésirables graves les plus fréquents rapportés chez ≥ 2 % des patients ont été le syndrome de relargage des cytokines (22,1 %), le sepsis (4,1 %), la COVID‑19 (3,4 %), la poussée tumorale (3,4 %), la pneumonie COVID‑19 (2,8 %), la neutropénie fébrile (2,1%), la neutropénie (2,1%) et l’épanchement pleural (2,1%).
Columvi a été arrêté définitivement en raison d’un effet indésirable chez 5,5 % des patients. Les effets indésirables les plus fréquents ayant conduit à l'arrêt définitif de Columvi ont été la COVID‑19 (1,4 %) et la neutropénie (1,4 %).
Columvi en association avec la gemcitabine et l’oxaliplatine
Les effets indésirables les plus fréquents (≥ 20 %) ont été la thrombopénie, le syndrome de relargage des cytokines, la neutropénie, l’anémie, les nausées, la neuropathie périphérique, la diarrhée, l’aspartate aminotransférase augmentée, l’alanine aminotransférase augmentée, le rash, la lymphopénie, la fièvre et les vomissements.
Les effets indésirables graves les plus fréquents rapportés chez ≥ 2 % des patients ont été le syndrome de relargage des cytokines (20,3 %), la fièvre (6,4 %), la pneumonie (5,8 %), la COVID-19 (5,8 %), la thrombopénie (4,7 %), l’infection des voies respiratoires (3,5 %), le sepsis (2,3 %), la neutropénie fébrile (2,3 %) et la diarrhée (2,3 %).
Columvi a été arrêté définitivement en raison d’un effet indésirable chez 20,9 % des patients. Les effets indésirables les plus fréquents ayant conduit à l’arrêt définitif de Columvi ont été la COVID-19 (11,6 %), le sepsis (1,2 %) et la pneumopathie inflammatoire (1,2 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables survenus chez des patients atteints d’un LDGCB réfractaire ou en rechute traités par Columvi en monothérapie (n = 145) dans l'étude NP30179 sont présentés dans le Tableau 6. Les patients ont reçu une médiane de 5 cycles de traitement par Columvi (intervalle : 1 à 13 cycles).
Les effets indésirables survenus chez des patients atteints d’un LDGCB réfractaire ou en rechute traités par Columvi en association avec la gemcitabine et l’oxaliplatine (n = 172) dans l’étude GO41944 (STARGLO) sont présentés dans le Tableau 7. Les patients ont reçu une médiane de 11 cycles de traitement par Columvi (intervalle : 1 à 13 cycles).
Les effets indésirables sont présentés par classe de systèmes d’organes MedDRA et par catégorie de fréquence. Les catégories de fréquence suivantes sont utilisées : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 6. Effets indésirables rapportés chez des patients atteints d’un LDGCB réfractaire ou en rechute traités par Columvi en monothérapie
Classe de systèmes d’organes | Effet indésirable | Tous les grades | Grade 34 |
Infections et infestations | Infections virales1 | Très fréquent | Fréquent* |
Infections bactériennes2 | Fréquent | Fréquent | |
Infections des voies respiratoires supérieures3 | Fréquent | Très rare** | |
Sepsis4 | Fréquent | Fréquent* | |
Infections des voies respiratoires inférieures5 | Fréquent | Très rare** | |
Pneumonie | Fréquent | Peu fréquent | |
Infection des voies urinaires6 | Fréquent | Peu fréquent | |
Infections fongiques7 | Fréquent | Très rare** | |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) | Poussée tumorale | Très fréquent | Fréquent |
Affections hématologiques et du système lymphatique | Neutropénie | Très fréquent | Très fréquent |
Anémie | Très fréquent | Fréquent | |
Thrombopénie | Très fréquent | Fréquent | |
Lymphopénie | Fréquent | Fréquent | |
Neutropénie fébrile8 | Fréquent | Fréquent | |
Affections du système immunitaire | Syndrome de relargage des cytokines9 | Très fréquent | |
Troubles du métabolisme et de la nutrition | Hypophosphatémie | Très fréquent | Fréquent |
Hypomagnésémie | Très fréquent | Très rare** | |
Hypocalcémie | Très fréquent | Très rare** | |
Hypokaliémie | Très fréquent | Peu fréquent | |
Hyponatrémie | Fréquent | Fréquent | |
Syndrome de lyse tumorale | Fréquent | Fréquent | |
Affections psychiatriques | Etat confusionnel | Fréquent | Très rare** |
Affections du système nerveux | Céphalée | Très fréquent | Très rare** |
Syndrome de neurotoxicité associé aux cellules effectrices immunitaires10 | Fréquent | Peu fréquent* | |
Somnolence | Fréquent | Peu fréquent | |
Tremblements | Fréquent | Très rare** | |
Myélite11 | Peu fréquent | Peu fréquent | |
Affections gastro-intestinales | Constipation | Très fréquent | Très rare** |
Diarrhée | Très fréquent | Très rare** | |
Nausées | Très fréquent | Très rare** | |
Hémorragie gastro-intestinale12 | Fréquent | Fréquent | |
Vomissements | Fréquent | Très rare** | |
Affections de la peau et du tissu sous-cutané | Rash13 | Très fréquent | Fréquent |
Troubles généraux et anomalies au site d’administration | Fièvre | Très fréquent | Très rare** |
Investigations | Alanine aminotransférase augmentée | Fréquent | Fréquent |
Aspartate aminotransférase augmentée | Fréquent | Fréquent | |
Phosphatases alcalines sanguines augmentées | Fréquent | Fréquent | |
Gamma-glutamyltransférase augmentée | Fréquent | Fréquent | |
Bilirubine sanguine augmentée | Fréquent | Peu Fréquent | |
Enzymes hépatiques augmentées | Fréquent | Fréquent |
* Des réactions de Grade 5 ont été rapportées. Voir Description d’effets indésirables sélectionnés.
** Aucun événement de Grade 3-4 n’a été rapporté.
1 Inclut : COVID‑19, pneumonie liée à la COVID‑19, zona, grippe et zona ophtalmique.
2 Inclut : infection de dispositif vasculaire, infection bactérienne, infection à Campylobacter, infection bactérienne des voies biliaires, infection bactérienne des voies urinaires, infection à Clostridium difficile, infection à Escherichia et péritonite.
3 Inclut : infection des voies respiratoires supérieures, sinusite, rhinopharyngite, sinusite chronique et rhinite.
4 Inclut : sepsis et choc septique.
5 Inclut : infection des voies respiratoires inférieures et bronchite.
6 Inclut : infection des voies urinaires et infection des voies urinaires à Escherichia.
7 Inclut : candidose œsophagienne et candidose buccale.
8 Inclut : neutropénie fébrile et infection neutropénique.
9 Basé sur la classification de consensus de l’ASTCT (Lee 2019).
10 ICANS basé sur Lee 2019 et se traduisant notamment par les symptômes suivants: somnolence, troubles cognitifs, état confusionnel, délire et désorientation.
11 Une myélite est survenue simultanément avec le SRC.
12 Inclut : hémorragie gastro-intestinale, hémorragie du gros intestin et hémorragie gastrique.
13 Inclut : rash, rash prurigineux, rash maculo-papuleux, dermatite, dermatite acnéiforme, dermatite exfoliative, érythème, érythème palmaire, prurit et rash érythémateux.
Tableau 7. Effets indésirables rapportés chez des patients atteints d’un LDGCB réfractaire ou en rechute traités par Columvi en association avec la gemcitabine et l’oxaliplatine
Classe de systèmes d’organes | Effet indésirable | Tous les grades | Grade 3–4 |
Infections et infestations | COVID-191 | Très fréquent | Fréquent* |
Infections des voies respiratoires2 | Très fréquent | Fréquent* | |
Pneumonie3 | Très fréquent | Fréquent* | |
Infections à cytomégalovirus4 | Fréquent | Peu fréquent | |
Infections herpétiques5 | Fréquent | Peu fréquent | |
Infection des voies urinaires6 | Fréquent | Fréquent | |
Sepsis7 | Fréquent | Fréquent* | |
Infections à Candida8 | Fréquent | Très rare** | |
Pneumonie à Pneumocystis jirovecii | Peu fréquent | Peu fréquent | |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) | Poussée tumorale9 | Fréquent | Très rare** |
Affections hématologiques et du système lymphatique | Thrombopénie | Très fréquent | Très fréquent |
Neutropénie | Très fréquent | Très fréquent | |
Anémie | Très fréquent | Très fréquent | |
Lymphopénie | Très fréquent | Très fréquent | |
Neutropénie fébrile | Fréquent | Fréquent | |
Affections du système immunitaire | Syndrome de relargage des cytokines10 | Très fréquent | Fréquent |
Troubles du métabolisme et de la nutrition | Hypokaliémie | Très fréquent | Fréquent |
Hyponatrémie | Très fréquent | Peu fréquent | |
Hypomagnésémie | Fréquent | Très rare** | |
Hypocalcémie | Fréquent | Peu fréquent | |
Hypophosphatémie | Fréquent | Fréquent | |
Syndrome de lyse tumorale | Fréquent | Fréquent | |
Affections du système nerveux | Neuropathie périphérique11 | Très fréquent | Fréquent |
Syndrome de neurotoxicité associé aux cellules effectrices immunitaires12 | Fréquent | Peu fréquent | |
Céphalée | Fréquent | Très rare** | |
Tremblements | Peu fréquent | Très rare** | |
Affections respiratoires, thoraciques et médiastinales | Pneumopathie inflammatoire | Fréquent | Très rare*,** |
Affections gastro-intestinales | Nausées | Très fréquent | Peu fréquent |
Diarrhée | Très fréquent | Fréquent | |
Vomissements | Très fréquent | Peu fréquent | |
Douleur abdominale13 | Très fréquent | Fréquent | |
Constipation | Très fréquent | Très rare** | |
Colite14 | Fréquent | Fréquent | |
Pancréatite15 | Fréquent | Fréquent | |
Affections de la peau et du tissu sous-cutané | Rash16 | Très fréquent | Peu fréquent |
Affections musculosquelettiques et du tissu conjonctif | Douleurs musculosquelettiques17 | Très fréquent | Fréquent |
Troubles généraux et anomalies au site d’administration | Fièvre | Très fréquent | Peu fréquent |
Investigations | Aspartate aminotransférase augmentée | Très fréquent | Fréquent |
Alanine aminotransférase augmentée | Très fréquent | Fréquent | |
Phosphatases alcalines sanguines augmentées | Très fréquent | Peu fréquent | |
Gamma-glutamyltransférase augmentée | Très fréquent | Fréquent | |
Lactate déshydrogénase sanguine augmentée | Très fréquent | Très rare** | |
Bilirubine sanguine augmentée18 | Fréquent | Très rare** | |
Enzymes hépatiques augmentées | Peu fréquent | Très rare** |
* Des réactions de Grade 5 ont été rapportées. Voir Description d’effets indésirables sélectionnés.
** Aucun événement de Grade 3-4 n’a été rapporté.
1 Inclut : COVID-19, pneumonie liée à la COVID-19 et test SARS-CoV-2 positif.
2 Inclut : infection des voies respiratoires supérieures, infection des voies respiratoires inférieures, infection des voies respiratoires et infection bactérienne des voies respiratoires.
3 Inclut : pneumonie, pneumonie bactérienne et pneumonie à pneumocoque.
4 Infection nouvelle ou réactivation. Inclut : infection à cytomégalovirus, test cytomégalovirus positif, réactivation d’une infection à cytomégalovirus et virémie à cytomégalovirus.
5 Infection nouvelle ou réactivation. Inclut : zona et infection herpétique.
6 Inclut : infection des voies urinaires et sepsis urinaire.
7 Inclut : sepsis, sepsis streptococcique, choc septique et sepsis à entérocoque.
8 Inclut : candidose buccale et infection à Candida.
9 Inclut : poussée tumorale et douleur liée à la masse tumorale.
10 Basé sur la classification de consensus de l’ASTCT (American Society for Transplantation and Cellular Therapy) (Lee 2019).
11 Inclut : neuropathie périphérique, neuropathie périphérique sensitive, dysesthésie, paresthésie, hypoesthésie, neuropathie motrice périphérique et polyneuropathie.
12 Inclut : état confusionnel, délire et ICANS.
13 Inclut : douleur abdominale, gêne abdominale, douleur abdominale haute, douleur abdominale basse et douleur gastro-intestinale.
14 Inclut : colite, colite ischémique et entérocolite.
15 Inclut : pancréatite et pancréatite aiguë.
16 Inclut : rash, rash prurigineux, rash maculo-papuleux, érythème, prurit, rash érythémateux, urticaire et érythème polymorphe.
17 Inclut : arthralgie, douleurs musculosquelettiques, dorsalgie, douleur osseuse, myalgie, cervicalgie, extrémités douloureuses, douleurs musculosquelettiques du thorax et douleur thoracique non cardiaque.
18 Inclut : bilirubine sanguine augmentée et hyperbilirubinémie.
Description d’effets indésirables sélectionnés
Les descriptions ci-dessous reflètent les informations relatives aux effets indésirables significatifs observés avec Columvi en monothérapie et/ou en association. Les détails concernant les effets indésirables significatifs observés avec Columvi administré en association sont présentés séparément si des différences cliniquement pertinentes ont été constatées par rapport à Columvi en monothérapie.
Syndrome de relargage des cytokines
Columvi en monothérapie
Un SRC de tout grade (selon les critères de l’ASTCT) est survenu chez 67,6% des patients ayant reçu Columvi en monothérapie, un SRC de Grade 1 ayant été rapporté chez 50,3 % des patients, un SRC de Grade 2 chez 13,1 % des patients, un SRC de Grade 3 chez 2,8 % des patients et un SRC de Grade 4 chez 1,4 % des patients. Un SRC est survenu plus d’une fois chez 32,4 % (47/145) des patients ; 36/47 patients ont présenté plusieurs événements de SRC de Grade 1 uniquement. Aucun cas de SRC d’issue fatale n’a été observé. Le SRC a été résolu chez tous les patients excepté un. Un patient a arrêté le traitement en raison d’un SRC.
Chez les patients présentant un SRC, les manifestations les plus fréquentes étaient : fièvre (99,0 %), tachycardie (25,5 %), hypotension artérielle (23,5 %), frissons (14,3 %) et hypoxie (12,2 %). Les événements de Grade 3 ou plus associés au SRC étaient : hypotension artérielle (3, 1%), hypoxie (3,1 %), fièvre (2,0 %) et tachycardie (2,0 %).
Un SRC de tout grade est survenu chez 54,5 % des patients après la première dose de 2,5 mg de Columvi au Jour 8 du Cycle 1, avec un délai médian d'apparition (à partir du début de la perfusion) de 12,6 heures (intervalle : 5,2 à 50,8 heures) et une durée médiane de 31,8 heures (intervalle : 0,5 à 316,7 heures) ; chez 33,3 % des patients après la dose de 10 mg au Jour 15 du Cycle 1, avec un délai médian d'apparition de 26,8 heures (intervalle : 6,7 à 125,0 heures) et une durée médiane de 16,5 heures (intervalle : 0,3 à 109,2 heures) ; et chez 26,8 % des patients après la dose de 30 mg au Cycle 2, avec un délai médian d'apparition de 28,2 heures (intervalle : 15,0 à 44,2 heures) et une durée médiane de 18,9 heures (intervalle : 1,0 à 180,5 heures). Un SRC a été rapporté chez 0,9 % des patients au Cycle 3 et chez 2 % des patients au-delà du Cycle 3.
Un SRC de Grade 2 est survenu chez 12,4 % des patients après la première dose de Columvi (2,5 mg), avec un délai médian d'apparition de 9,7 heures (intervalle : 5,2 à 19,1 heures) et une durée médiane de 50,4 heures (intervalle : 6,5 à 316,7 heures). Après la dose de 10 mg de Columvi au Jour 15 du Cycle 1, l’incidence des SRC de Grade 2 a diminué à 5,2 % des patients, avec un délai médian d'apparition de 26,2 heures (intervalle : 6,7 à 144,2 heures) et une durée médiane de 30,9 heures (intervalle : 3,7 à 227,2 heures). Un SRC de Grade 2 après l’administration de Columvi à la dose de 30 mg au Jour 1 du Cycle 2 est survenu chez un patient (0,8 %), avec un délai d'apparition de 15,0 heures et une durée de 44,8 heures. Aucun SRC de Grade 2 n’a été rapporté au-delà du Cycle 2.
Chez 145 patients, 7 patients (4,8 %) ont présenté des tests de la fonction hépatique élevés (ASAT et ALAT > 3 x LSN et/ou bilirubine totale > 2 x LSN) rapportés de manière concomitante avec un SRC (n = 6) ou avec une progression de la maladie (n = 1).
Parmi les 25 patients ayant présenté un SRC de Grade 2 après l’administration de Columvi, 22 (88,0 %) ont reçu du tocilizumab, 15 (60,0 %) ont reçu des corticoïdes et 14 (56,0 %) ont reçu à la fois du tocilizumab et des corticoïdes. Dix patients (40,0 %) ont reçu de l’oxygène. L’ensemble des 6 patients (24,0 %) avec un SRC de Grade 3 ou 4 ont reçu un seul vasopresseur.
Des hospitalisations en raison de la survenue d’un SRC après l’administration de Columvi ont eu lieu chez 22,1 % des patients et la durée médiane de l’hospitalisation rapportée était de 4 jours (intervalle : 2 à 15 jours).
Columvi en association avec la gemcitabine et l’oxaliplatine
Un SRC de tout grade (selon les critères de l’ASTCT) est survenu chez 44,2 % des patients ayant reçu Columvi en association avec la gemcitabine et l’oxaliplatine, un SRC de Grade 1 ayant été rapporté chez 31,4 % des patients, un SRC de Grade 2 chez 10,5 % des patients et un SRC de Grade 3 chez 2,3 % des patients. Un SRC est survenu plus d’une fois chez 21,5 % (37/172) des patients ; 30/37 patients ont présenté plusieurs événements de SRC de Grade 1 uniquement. Aucun cas de SRC de Grade 4 ou d’issue fatale n’a été observé. Le SRC a été résolu chez tous les patients excepté un. Un patient a arrêté le traitement en raison d’un SRC.
Chez les patients présentant un SRC, les manifestations les plus fréquentes étaient : fièvre (98,7 %), hypotension (22,4 %), frissons (17,1 %) et hypoxie (14,5 %). Les événements de Grade 3 ou plus associés au SRC étaient : hypotension (6,6 %), hypoxie (5,3 %), fièvre (3,9 %), frissons (1,3 %) et diarrhée (1,3 %).
Un SRC de tout grade est survenu chez 34,9 % des patients après la première dose de 2,5 mg de Columvi au Jour 8 du Cycle 1, avec un délai médian d’apparition (à partir du début de la perfusion) de 12,6 heures (intervalle : 4,4 à 54,7 heures) et une durée médiane de 19,8 heures (intervalle : 2,0 à 168,0 heures) ; chez 14,4 % des patients après la dose de 10 mg au Jour 15 du Cycle 1, avec un délai médian d’apparition de 22,8 heures (intervalle : 7,4 à 81,2 heures) et une durée médiane de 10,6 heures (intervalle : 1,0 à 248,5 heures) ; et chez 9,3 % des patients après la dose de 30 mg au Cycle 2, avec un délai médian d’apparition de 23,5 heures (intervalle : 14,7 à 33,4 heures) et une durée médiane de 18,4 heures (intervalle : 8,3 à 137,0 heures). Un SRC a été rapporté chez 6,7 % des patients au Cycle 3 et chez 11,0 % des patients au-delà du Cycle 3.
Un SRC de Grade ≥ 2 est survenu chez 10,5 % des patients après la première dose de Columvi (2,5 mg), avec un délai médian d’apparition de 12,0 heures (intervalle : 4,4 à 30,5 heures) et une durée médiane de 42,3 heures (intervalle : 3,5 à 143,7 heures). La majorité (14/18) des patients ayant présenté un SRC de Grade ≥ 2 ont présenté un SRC dans les 8 heures suivant le début de la première dose de Columvi (2,5 mg). Après la dose de 10 mg de Columvi au Jour 15 du Cycle 1, l’incidence des SRC de Grade ≥ 2 a diminué à 1,8 % des patients, avec un délai médian d’apparition de 22,3 heures (intervalle : 7,4 à 22,8 heures) et une durée médiane de 37,0 heures (intervalle : 34,8 à 248,5 heures). Aucun événement de SRC de Grade ≥ 2 n’a été observé après l’administration de Columvi à la dose de 30 mg au Jour 1 du Cycle 2. Trois patients (2,0 %) ont présenté un SRC de Grade ≥ 2 au-delà du Cycle 2 (tous les événements étaient de Grade 2).
Sur les 172 patients, 2 patients (1,2 %) ont présenté des tests de la fonction hépatique élevés (ASAT et ALAT > 3 x LSN) rapportés de manière concomitante avec un SRC.
Sur les 76 patients ayant présenté un SRC de tout grade, 28 patients (36,8 %) ont été traités par le tocilizumab, 39 patients (51,3 %) ont été traités par des corticoïdes et 18 patients (23,7 %) ont reçu à la fois du tocilizumab et des corticoïdes.
Parmi les 22 patients ayant présenté un SRC de Grade ≥ 2 après l’administration de Columvi, 16 (72,7 %) ont reçu du tocilizumab, 15 (68,2 %) ont reçu des corticoïdes et 12 (54,5 %) ont reçu à la fois du tocilizumab et des corticoïdes. Onze patients (50,0 %) ont reçu de l’oxygène. L’ensemble des 4 patients (18,2 %) avec un SRC de Grade 3 ont reçu un seul vasopresseur.
Des hospitalisations en raison de la survenue d’un SRC après l’administration de Columvi ont eu lieu chez 19,8 % des patients et la durée médiane de l’hospitalisation rapportée était de 5 jours (intervalle : 2 à 85 jours).
Syndrome de neurotoxicité associé aux cellules effectrices immunitaires
Des cas d’ICANS, y compris de grade 3 et au-delà, ont été signalés lors d’essais cliniques et depuis la commercialisation. Les manifestations cliniques les plus fréquentes de l’ICANS étaient les suivantes : confusion, diminution du niveau de conscience, désorientation, crises convulsives, aphasie et dysgraphie. Sur la base des données disponibles, l’apparition d’une toxicité neurologique était concomitante à la survenue d’un SRC dans la plupart des cas.
Le délai d’apparition observé de la majorité des ICANS était de 1 à 7 jours, avec une médiane de 2 jours après la dose la plus récente. Seuls quelques événements ont été signalés plus d’un mois après le début du traitement par Columvi.
Infections graves
Des infections graves ont été rapportées chez 15,9 % des patients ayant reçu Columvi en monothérapie. Les infections graves les plus fréquentes rapportées chez ≥ 2 % des patients étaient les suivantes : sepsis (4,1 %), COVID‑19 (3,4 %) et pneumonie COVID‑19 (2,8 %). Des décès liés à une infection ont été rapportés chez 4,8 % des patients (dus à : sepsis, pneumonie COVID‑19 et COVID‑19). Quatre patients (2,8 %) ont présenté des infections graves de manière simultanée avec une neutropénie de Grade 3 ou 4.
Des infections graves ont été rapportées chez 22,7 % des patients ayant reçu Columvi en association avec la gemcitabine et l’oxaliplatine. Les infections graves les plus fréquentes rapportées chez ≥ 2 % des patients étaient les suivantes : pneumonie (5,8 %), COVID-19 (4,7 %) et infections des voies respiratoires inférieures (2,9 %). Des décès liés à une infection ont été rapportés chez 3,5 % des patients (dus à : COVID-19, pneumonie, infection des voies respiratoires et choc septique). Un patient (0,6 %) a présenté une infection grave (pneumonie) de manière simultanée avec une neutropénie de Grade 3.
Pneumopathie inflammatoire
Des événements de type pneumopathie inflammatoire (à l’exception des pneumonies d’étiologie infectieuse) ont été rapportés chez 2 patients (1,2 %) ayant reçu Columvi en association avec la gemcitabine et l’oxaliplatine, et ont tous deux été d’issue fatale. Le délai médian d’apparition de la pneumopathie inflammatoire à partir de la première dose de Columvi a été de 168 jours (intervalle : 102 à 255 jours).
Colite
Des événements de type colite (à l’exception d’une étiologie infectieuse) ont été rapportés chez 4 des 172 patients (2,3 %) ayant reçu Columvi en association avec la gemcitabine et l’oxaliplatine. Deux patients (1,2 %) ont présenté des événements de Grade 3. Le délai médian d’apparition de la colite à partir de la première dose de Columvi a été de 154 jours (intervalle : 115 à 187 jours).
Infections opportunistes
Des événements à cytomégalovirus (CMV) ont été rapportés chez 10 patients (5,8 %) ayant reçu Columvi en association avec la gemcitabine et l’oxaliplatine, 1 patient (0,6 %) ayant présenté une virémie à CMV de Grade 3. Une candidose buccale a été rapportée chez 3 patients (1,7 %), tous ayant présenté des événements de Grade 1 à 2. Une pneumonie à Pneumocystis jirovecii (Grade 3) a été rapportée chez 1 patient (0,6 %), le même patient présentant une virémie à CMV de Grade 3. Une méningite à Borrelia (Grade 2) a été rapportée chez 1 patient (0,6 %).
Neutropénie
Des cas de neutropénie (incluant une diminution du nombre de neutrophiles) ont été rapportés chez 40% des patients et des cas de neutropénie sévère (Grade 3 ou 4) chez 29% des patients ayant reçu Columvi en monothérapie. Le délai médian d’apparition de la première neutropénie était de de 29 jours (intervalle : 1 à 203 jours). Une neutropénie prolongée (durant plus de 30 jours) est survenue chez 11,7 % des patients. La majorité des patients présentant une neutropénie (79,3 %) étaient traités par G‑CSF. Une neutropénie fébrile a été rapportée chez 3,4 % des patients.
Poussée tumorale
Des cas de poussée tumorale ont été rapportés chez 11,7 % des patients ayant reçu Columvi en monothérapie, avec une poussée tumorale de Grade 2 chez 4,8 % des patients et une poussée tumorale de Grade 3 chez 2,8 % des patients. Il a été observé une poussée tumorale impliquant des ganglions lymphatiques de la tête et du cou et se traduisant par une douleur, et une poussée tumorale impliquant des ganglions lymphatiques du thorax avec des symptômes de type essoufflement dus au développement d’un épanchement pleural. La plupart des événements de poussée tumorale (16/17) sont survenus pendant le Cycle 1 et aucun événement de ce type n’a été rapporté au-delà du Cycle 2. Le délai médian d'apparition de la poussée tumorale de tout grade était de 2 jours (intervalle : 1 à 16 jours) et la durée médiane était de 3,5 jours (intervalle : 1 à 35 jours).
Parmi les 11 patients ayant présenté une poussée tumorale de Grade ≥ 2, 2 patients (18,2 %) ont reçu des antalgiques, 6 patients (54,5 %) ont reçu des corticoïdes et des antalgiques incluant des dérivés morphiniques, 1 patient (9,1 %) a reçu des corticoïdes et des antiémétiques et 2 patients (18,2 %) n’ont pas nécessité de traitement. Tous les événements de poussée tumorale ont été résolus, sauf chez un patient ayant présenté un événement de Grade ≥ 2. Aucun patient n’a arrêté le traitement en raison d’une poussée tumorale.
Syndrome de lyse tumorale
Un SLT a été rapporté chez 2 patients (1,4 %) ayant reçu Columvi en monothérapie ; il était de Grade 3 dans les deux cas. La durée médiane d'apparition du SLT était de 2 jours et la durée médiane était de 4 jours (intervalle : 3 à 5 jours).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil‑Barell‑Strasse 1
79639 Grenzach‑Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/23/1742/001
EU/1/23/1742/002
10. DATE DE MISE À JOUR DU TEXTE
8 mai 2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4729232 | COLUMVI 2,5MG SOL DIL PERF FL INJ 2,5ML | - | € 774 | Oui | - | - | |
| 4729240 | COLUMVI 10MG SOL DIL PERF FL INJ 10ML | - | € 3095 | Oui | - | - |