RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1 DÉNOMINATION DU MÉDICAMENT
Piasky 340 mg solution injectable/pour perfusion
2 COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon de 2 mL contient 340 mg de crovalimab.
Chaque mL de solution injectable/pour perfusion contient 170 mg de crovalimab.
Le crovalimab est un anticorps monoclonal humanisé produit sur cellules ovariennes de hamster chinois (CHO) par la technique de l’ADN recombinant.
Pour la liste complète des excipients, voir rubrique 6.1.
3 FORME PHARMACEUTIQUE
Solution injectable/pour perfusion (injection/perfusion).
Solution limpide à fortement opalescente et presque incolore à jaune brunâtre. La solution a un pH d’environ 5,8 et une osmolalité d’environ 297 mOsm/kg.
4 INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Piasky est indiqué en monothérapie pour le traitement des patients adultes et pédiatriques âgés de 12 ans et plus, pesant 40 kg et plus, atteints d’hémoglobinurie paroxystique nocturne (HPN) :
Chez les patients présentant une hémolyse avec un ou des symptômes cliniques indiquant une forte activité de la maladie.
Chez les patients qui sont cliniquement stables après avoir été traités par un inhibiteur de la fraction C5 du complément pendant au moins les 6 derniers mois.
4.2 Posologie et mode d’administration
Le traitement doit être instauré sous la surveillance d’un médecin expérimenté dans le traitement des troubles hématologiques.
Posologie
Le schéma posologique recommandé consiste en une dose de charge administrée par perfusion intraveineuse (le jour 1), suivie de quatre doses de charge hebdomadaires supplémentaires administrées par injection sous‑cutanée (les jours 2, 8, 15 et 22). La dose d’entretien commence le jour 29 et est ensuite administrée toutes les 4 semaines par injection sous‑cutanée. Les doses à administrer sont basées sur le poids corporel du patient, comme indiqué dans le Tableau 1.
Pour les patients précédemment traités par un autre inhibiteur du complément, la première dose de charge intraveineuse de Piasky doit être administrée au moment de l’administration suivante prévue de l’inhibiteur du complément (voir rubrique 4.4 pour des informations supplémentaires concernant le passage à un autre inhibiteur de la fraction C5 du complément). L’administration par voie sous‑cutanée des doses de charge supplémentaires et des doses d’entretien de Piasky se fera selon le schéma présenté dans le Tableau 1.
Tableau 1 : Schéma posologique de Piasky en fonction du poids corporel
Poids corporel | ≥ 40 kg à < 100 kg | ≥ 100 kg |
Dose de charge | | |
Dose d’entretien | | |
Le schéma d’administration peut occasionnellement varier de ± 2 jours par rapport au jour d’administration prévu (sauf le jour 1 et le jour 2). Dans ce cas, la dose suivante doit être administrée selon le schéma habituel.
Durée du traitement
Piasky est destiné à un traitement au long cours, sauf si l’arrêt de ce médicament est cliniquement indiqué (voir rubrique 4.4).
Retard ou omission de prise
En cas d’oubli d’une dose entière ou partielle prévue de Piasky, la dose oubliée ou le restant de la dose prévue doit être administrée le plus tôt possible avant le jour de la prochaine dose programmée. La dose suivante doit ensuite être administrée le jour prévu. Ne prenez pas deux doses ou n’administrez pas plus que la dose prescrite le même jour pour compenser la dose manquée.
Modifications de la posologie
Une modification de la dose d’entretien est nécessaire si le poids corporel du patient change de 10 % ou plus et se stabilise au-dessus ou en dessous de 100 kg au cours du traitement (voir Tableau 1 pour la dose recommandée). En conséquence, le poids corporel du patient doit être surveillé périodiquement et de façon continue, le cas échéant.
Populations particulières
Personnes âgées
Aucun ajustement posologique n’est nécessaire chez les patients âgés de 65 ans et plus, bien que l’expérience avec crovalimab chez les patients âgés dans les études cliniques soit limitée (voir rubrique 5.2).
Insuffisance rénale
Aucun ajustement posologique n’est recommandé chez les patients présentant une insuffisance rénale légère, modérée ou sévère (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère. Crovalimab n’a pas été étudié chez les patients présentant une insuffisance hépatique modérée à sévère et aucune recommandation sur la posologie ne peut être donnée (voir rubrique 5.2).
Population pédiatrique
Aucun ajustement posologique de crovalimab n’est nécessaire chez les patients pédiatriques âgés de 12 ans et plus, ayant un poids corporel ≥ 40 kg. La tolérance et l’efficacité de crovalimab chez les enfants âgés de moins de 12 ans et chez les enfants pesant < 40 kg n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
Piasky est administré en perfusion intraveineuse (première dose) et en injection sous‑cutanée (doses suivantes).
Administration intraveineuse
Piasky doit être préparé pour une administration intraveineuse en utilisant une technique aseptique appropriée. Piasky doit être dilué et administré par un professionnel de santé par perfusion intraveineuse pendant 60 minutes ± 10 minutes (1 000 mg) ou 90 minutes ± 10 minutes (1 500 mg). Piasky ne doit pas être administré en injection rapide ou bolus intraveineux.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique 6.6.
La perfusion de crovalimab peut être ralentie ou interrompue si le patient développe une réaction liée à la perfusion. La perfusion doit être immédiatement interrompue si le patient présente une réaction d’hypersensibilité grave (voir rubrique 4.4).
Administration sous‑cutanée
Piasky doit être utilisé non dilué et doit être préparé en utilisant une technique aseptique appropriée. Il est recommandé d’injecter Piasky dans l’abdomen. Dans l’abdomen, les sites d’injection doivent être alternés à chaque injection. Les injections ne doivent jamais être réalisées dans des grains de beauté, des cicatrices ou des zones où la peau est sensible, contusionnée, rouge, durcie ou lésée.
Administration par le patient et/ou un aidant
Après avoir reçu une formation appropriée à la technique d’injection sous‑cutanée, le patient peut s’auto-administrer Piasky ou l’aidant peut administrer Piasky sans la supervision d’un professionnel de santé, si le médecin prescripteur estime que cela est approprié.
Des instructions détaillées sur l’administration de Piasky sont fournies à la fin de la Notice.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Patients présentant une infection à Neisseria meningitidis non résolue.
Patients qui ne sont pas actuellement vaccinés contre Neisseria meningitidis, à moins qu’ils ne reçoivent une antibioprophylaxie appropriée jusqu’à 2 semaines après la vaccination (voir rubrique 4.4).
4.8 Effets indésirables
Résumé du profil de tolérance
Les effets indésirables les plus fréquents observés étaient les réactions à complexes immuns de type III (18,9 % chez les patients qui sont passés d’un traitement avec un autre inhibiteur du C5 à crovalimab) les infections des voies aériennes supérieures (18,6 %), la fièvre (13,5 %), les céphalées (10,9 %) et les réactions liées à la perfusion (10,2 %). Les effets indésirables graves les plus fréquents observés étaient des réactions à complexes immuns de type III (4,0 % chez les patients qui sont passés d’un traitement avec un autre inhibiteur de C5 à crovalimab) et des pneumonies (1,5 %).
Les données de tolérance issues des 44 patients de l’étude COMPOSER, chez lesquels la durée médiane de traitement était de 4,69 ans (intervalle : 0,4 – 6,3 ans), n’ont pas révélé de problèmes de sécurité supplémentaires liés à l’utilisation prolongée de crovalimab.
Tableau des effets indésirables
La tolérance de crovalimab chez les patients atteints d’HPN a été évaluée dans trois études de phase III, COMMODORE 2 (BO42162), COMMODORE 3 (YO42311) et COMMODORE 1 (BO42161), ainsi que dans une étude de phase I/II, COMPOSER (BP39144).
Le Tableau 2 liste les effets indésirables qui ont été rapportés lors de l’utilisation de crovalimab dans une analyse groupée des 393 patients inclus dans les études de phase III, sauf mention contraire. La durée médiane du traitement par crovalimab basée sur l’analyse groupée des 393 patients était de 64 semaines (intervalle : 0,1 – 136,4 semaines).
Les effets indésirables sont présentés par classes de systèmes d’organes MedDRA. La catégorie de fréquence correspondante pour chaque effet indésirable est basée sur la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000). Dans chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 2 : Résumé des effets indésirables survenus chez les patients traités par Piasky
Classe de systèmes d’organes MedDRA | Effets indésirables (MedDRA) | Catégorie de fréquence |
Infections et infestations | Infection des voies aériennes supérieures | Très fréquent |
Pneumonie | Fréquent | |
Infection des voies aériennes | ||
Infection urinaire | ||
Rhinopharyngite | ||
Septicémie | Peu fréquent | |
Choc septique | ||
Bactériémie | ||
Pyélonéphrite | ||
Affections du système immunitaire | Réaction à complexes immuns de type III* | Très fréquent |
Hypersensibilité | Fréquent | |
Affections du système nerveux | Céphalées | Très fréquent |
Affections gastro-intestinales | Douleur abdominale | Fréquent |
Diarrhée | ||
Affections de la peau et du tissu sous-cutané | Éruption cutanée | Fréquent |
Affections musculo-squelettiques et du tissu conjonctif | Arthralgies | Fréquent |
Troubles généraux et anomalies au site d’administration | Fièvre | Très fréquent |
Asthénie | Fréquent | |
Fatigue | ||
Réaction au site d’injection | Peu fréquent | |
Lésions, intoxications et complications liées aux procédures | Réaction liée à la perfusion | Très fréquent |
Réaction liée à l’injection | Fréquent |
*La réaction à complexes immuns de type III (également appelée réaction à complexes immuns de type III) est limitée aux patients qui passent d’un autre inhibiteur de C5 au crovalimab ou de crovalimab à un autre inhibiteur de C5. La fréquence des réactions à complexes immuns de type III est rapportée pour un sous‑groupe de N = 201 patients qui sont passés d’un traitement avec un autre inhibiteur de C5 au crovalimab, avec des taux d’incidence calculés en prenant ces N = 201 patients comme dénominateur. Voir ci-dessous.
Description de certains effets indésirables
Réactions à complexes immuns de type III (voir rubriques 4.4 et 4.5)
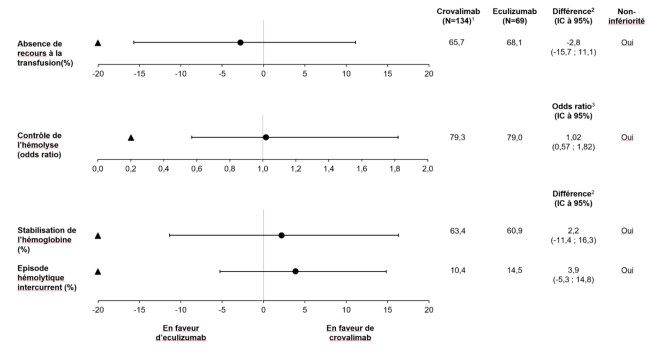
Dans les études de phase III, 19,4 % (39 sur 201) des patients qui sont passés d’un traitement par eculizumab ou ravulizumab à crovalimab ont présenté une réaction à complexes immuns de type III (rapportée comme une réaction médiée par complexes immuns de type III). Parmi ces 39 patients, 2 patients ont présenté une seconde réaction à complexes immuns de type III après l’arrêt de crovalimab et le passage au ravulizumab. Les signes et symptômes les plus fréquemment rapportés étaient des arthralgies et des éruptions cutanées, et les autres symptômes rapportés comprennent : fièvre, céphalées, myalgies, douleurs abdominales, asthénie/fatigue et neuropathie axonale. Le délai médian de survenue d’une réaction à complexes immuns de type III chez les patients qui sont passés d’un traitement par eculizumab ou ravulizumab à crovalimab a été de 1,6 semaine (intervalle : 0,7 – 4,4 semaines), et 5,1 % des patients (2 sur 39) ont présenté une réaction à complexes immuns de type III avec un délai de survenue supérieur à 4 semaines. La plupart des cas de réaction à complexes immuns de type III étaient transitoires et d’une durée médiane de 1,7 semaine (intervalle : 0,4 – 34,1 semaines). Parmi les patients traités par crovalimab précédemment traités par eculizumab ou ravulizumab, la majorité a présenté un événement de grade 1 ou 2 (23 sur 39 patients), 8 % (16 sur 39) ont présenté des événements de grade 3. La plupart des événements ont été résolus sans modification du traitement par crovalimab.
Dans l’étude COMPOSER, parmi les 26 patients passés d’eculizumab au crovalimab, 2 patients ont rapporté chacun 1 événement indésirable de réaction à complexes immuns de type III. Ces événements étaient légers/modérés et non graves. Un autre patient a développé une légère réaction à complexes immuns de type III après avoir arrêté crovalimab et être passé à un autre inhibiteur de C5.
Immunogénicité
Dans deux études randomisées de phase III (COMMODORE 1 et COMMODORE 2) et une étude de phase III à un seul bras (COMMODORE 3), le statut d’ADA était évaluable chez 392 patients. Sur ces 392 patients, 118 (30,1 %) étaient ADA-positifs. Aucune différence n’a été observée au niveau des taux d’effets indésirables généralement associés à l’immunogénicité (telles que les réactions liées à la perfusion, les réactions au site d’injection ou l’hypersensibilité) entre les patients ADA-positifs et les patients ADA-négatifs (voir rubrique 5.1).
Immunogénicité entraînant une exposition moindre et une perte d’efficacité
Les patients peuvent développer des ADA susceptibles d’interférer avec l’exposition au crovalimab. Sur les 392 patients dont le statut ADA a été évalué, une diminution partielle ou totale de l’exposition associée à l’apparition des ADA a été observée chez 23 patients (5,9 %) ; parmi eux, 17 (4,3 %) ont présenté une perte d’activité pharmacologique coïncidant avec une diminution d’exposition et une perte d’efficacité, se manifestant par une perte durable du contrôle de l’hémolyse chez 7 patients (1,8 %).
En cas de signes cliniques de perte d’efficacité, une évaluation rapide par un professionnel de santé doit être envisagée (voir rubrique 4.4).
Réactions aux sites de perfusion et d’injection
Dans les études de phase III, 10,2 % des patients traités par crovalimab ont présenté une réaction liée à la perfusion. Les signes et symptômes les plus fréquemment rapportés étaient les suivants : céphalées (7,1 %), éruption cutanée (0,8 %), sensation vertigineuse (0,8 %), douleur abdominale (0,5 %), érythème (0,5 %), nausée (0,5 %), fièvre (0,5 %) et paresthésie (0,3 %). Tous les événements rapportés étaient de grade 1 à 2.
Dans les études de phase III, 8,4 % des patients traités par crovalimab ont présenté une réaction liée à l’injection. Les signes et symptômes les plus fréquemment rapportés étaient les suivants : céphalées (2,5 %), érythème au site d’injection (1,0 %), douleur au site d’injection (1,0 %) et éruption cutanée au site d’injection (1,0 %). La majorité des événements était de grade 1 ou 2.
Infections par des bactéries encapsulées
Compte tenu de son mécanisme d’action, l’utilisation de crovalimab pourrait augmenter le risque d’infections, en particulier les infections causées par des bactéries encapsulées dont Streptococcus pneumoniae, Neisseria meningitidis de types A, C, W, Y et B et Haemophilus influenzae (voir rubrique 4.4).
Dans les études de phase III, les infections à bactéries encapsulées qui ont été rapportées étaient Klebsiella pneumoniae, Klebsiella (non spécifié), Haemophilus influenzae et Neisseria subflava, cette dernière ayant entraîné un événement indésirable de bactériémie chez un patient.
Population pédiatrique
Chez 12 patients pédiatriques atteints d’HPN avec un poids corporel ≥ 40 kg (âgés de 13 à 17 ans) inclus dans les études COMMODORE 1, COMMODORE 2 et COMMODORE 3, la tolérance est apparue similaire à celle observée chez les patients adultes atteints d’HPN. Les effets indésirables associés au crovalimab qui ont été rapportés chez les patients pédiatriques atteints d’HPN sont les suivants : infection des voies aériennes supérieures (16,7 %), infection urinaire (16,7 %), fatigue (16,7 %), fièvre (16,7 %), céphalées (8,3 %), réaction liée à la perfusion (8,3 %) et réaction liée à l’injection (8,3 %).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4866901 | Piasky 340 mg solution injectable/pour perfusion | - | € 11213,29 | Oui | - | - |