RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Tecentriq 840 mg, solution à diluer pour perfusion
Tecentriq 1 200 mg, solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Tecentriq 840 mg, solution à diluer pour perfusion
Un flacon de 14 mL de solution à diluer contient 840 mg d’atezolizumab*
Tecentriq 1 200 mg, solution à diluer pour perfusion
Un flacon de 20 mL de solution à diluer contient 1 200 mg d’atezolizumab*
Après dilution (voir rubrique 6.6), la concentration finale de la solution diluée doit être entre 3,2 et 16,8 mg/mL.
*L'atezolizumab est un anticorps monoclonal humanisé anti-PD-L1 (Programmed Death-Ligand 1) de type IgG1, à Fc modifié, produit dans des cellules d'ovaire de hamster chinois par la technique de l'ADN recombinant.
Excipient à effet notoire
Chaque flacon de 840 mg de Tecentriq contient 5,6 mg de polysorbate 20.
Chaque flacon de 1 200 mg de Tecentriq contient 8 mg de polysorbate 20.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution à diluer pour perfusion.
Liquide limpide, incolore à légèrement jaunâtre. La solution a un pH de 5,5 - 6,1 et une osmolalité de 129 - 229 mOsm/kg.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
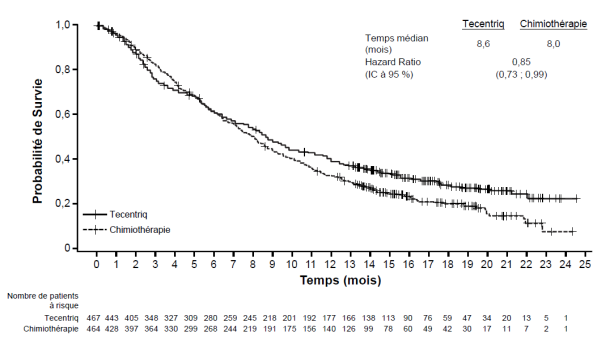
Carcinome urothélial
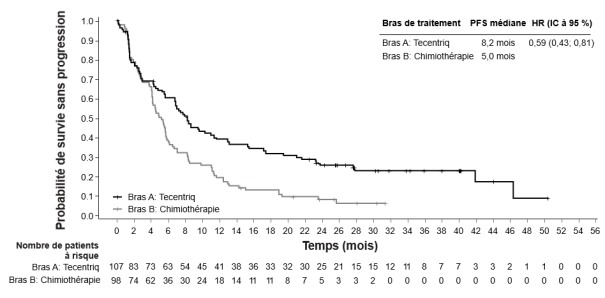
Tecentriq en monothérapie est indiqué dans le traitement des patients adultes atteints d'un carcinome urothélial localement avancé ou métastatique :
après une chimiothérapie antérieure à base de platine, ou
considérés inéligibles au cisplatine et dont les tumeurs présentent une expression de PD-L1
≥ 5 % (voir rubrique 5.1).
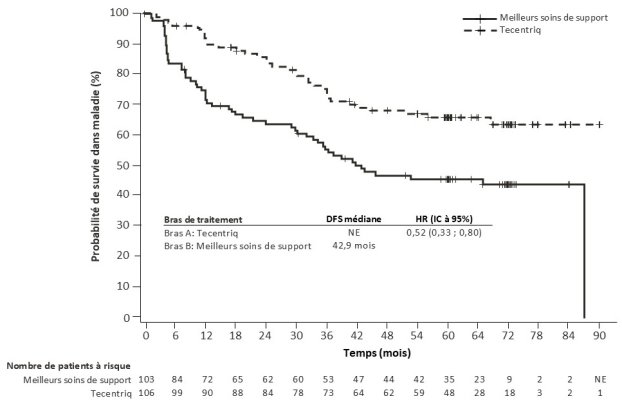
Cancer bronchique non à petites cellules (CBNPC) de stade précoce
Tecentriq en monothérapie est indiqué dans le traitement adjuvant, après résection complète et chimiothérapie à base de platine, des patients adultes atteints d’un cancer bronchique non à petites cellules (CBNPC) avec un risque élevé de récidive, dont les tumeurs présentent une expression de
PD-L1 ≥ 50 % sur les cellules tumorales (TC) et qui ne présentent pas de CBNPC avec EGFR muté ou réarrangement du gène ALK (ALK-positif) (voir rubrique 5.1 pour les critères de sélection).
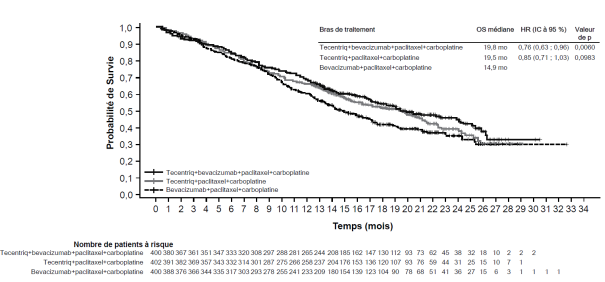
CBNPC avancé
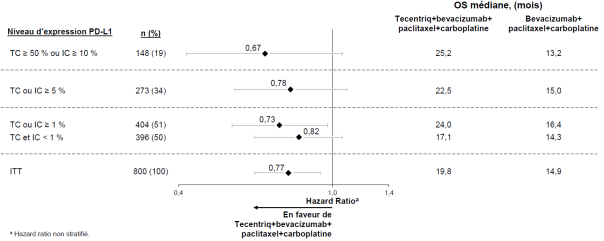
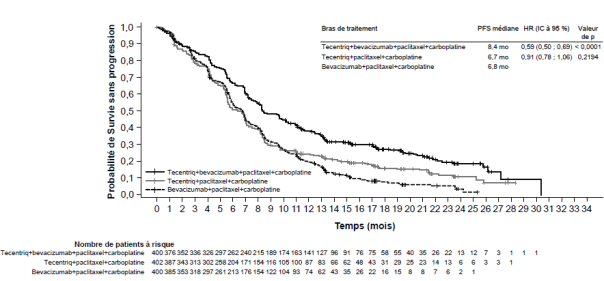
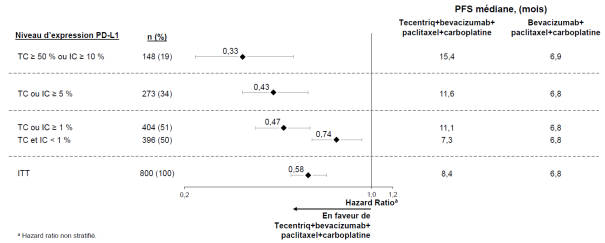
Tecentriq, en association au bevacizumab, paclitaxel et carboplatine, est indiqué en première ligne de traitement des patients adultes atteints d’un CBNPC non épidermoïde métastatique. Chez les patients atteints d’un CBNPC avec EGFR muté ou réarrangement du gène ALK (ALK-positif), Tecentriq, en association au bevacizumab, paclitaxel et carboplatine, est indiqué seulement après échec des thérapies ciblées appropriées (voir rubrique 5.1).
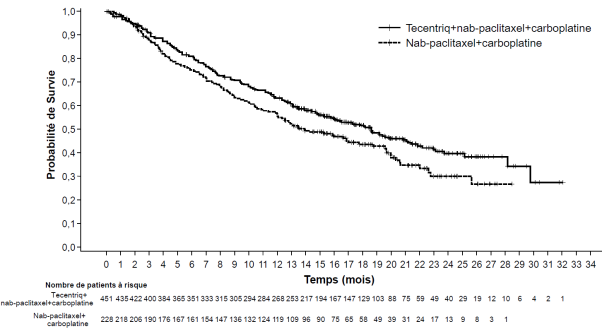
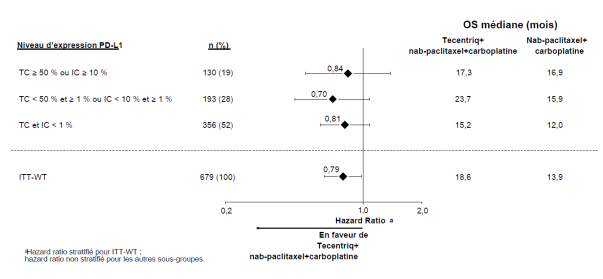
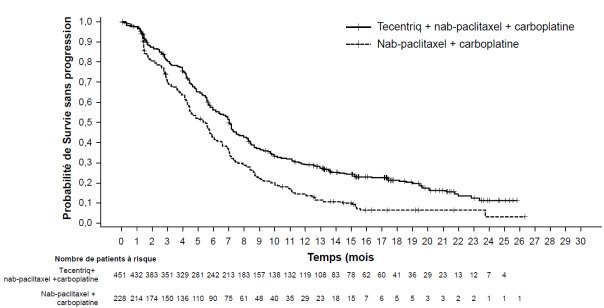
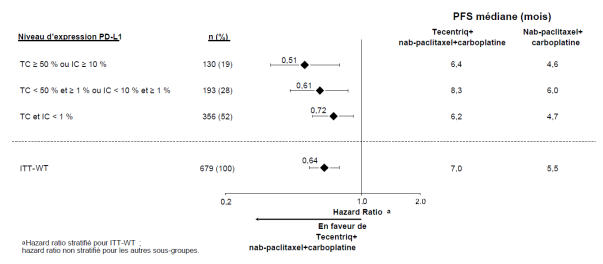
Tecentriq, en association au nab-paclitaxel et carboplatine, est indiqué en première ligne de traitement des patients adultes atteints d’un CBNPC non épidermoïde métastatique sans EGFR muté ou réarrangement du gène ALK (ALK-positif) (voir rubrique 5.1).
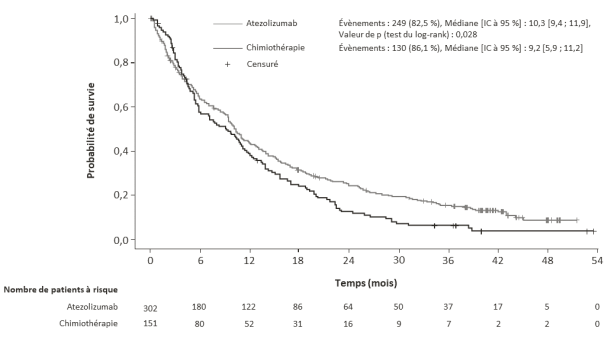
Tecentriq en monothérapie est indiqué dans le traitement de première ligne des patients adultes atteints d’un CBNPC métastatique dont les tumeurs présentent une expression de PD-L1 ≥ 50 % sur les TC ou ≥ 10 % sur les cellules immunitaires infiltrant la tumeur (IC) et qui ne sont pas atteints d’un CBNPC avec EGFR muté ou réarrangement du gène ALK (ALK-positif) (voir rubrique 5.1).
Tecentriq en monothérapie est indiqué dans le traitement de première ligne des patients adultes atteints d’un CBNPC avancé inéligibles à un traitement à base de sels de platine (voir rubrique 5.1 pour les critères de sélection).
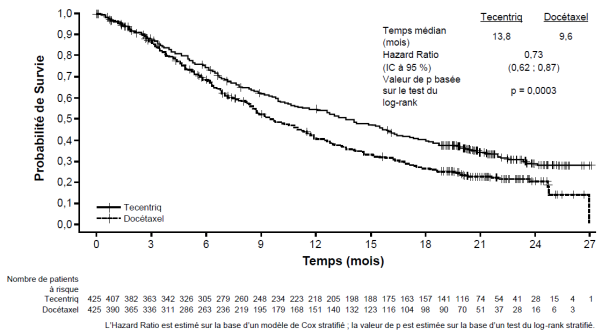
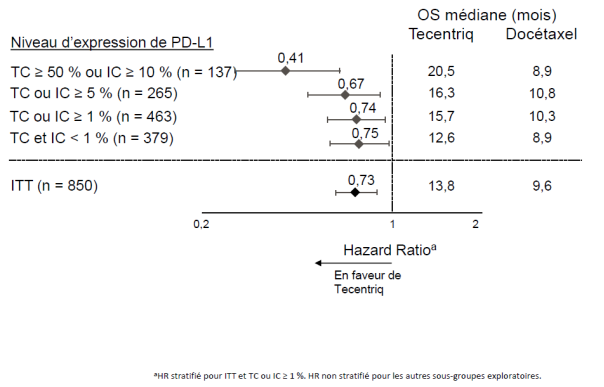
Tecentriq en monothérapie est indiqué dans le traitement des patients adultes atteints d'un CBNPC localement avancé ou métastatique après une chimiothérapie antérieure. Les patients atteints d’un CBNPC avec EGFR muté ou réarrangement du gène ALK (ALK-positif) doivent également avoir reçu des thérapies ciblées avant de recevoir Tecentriq (voir rubrique 5.1).
Cancer bronchique à petites cellules (CBPC)
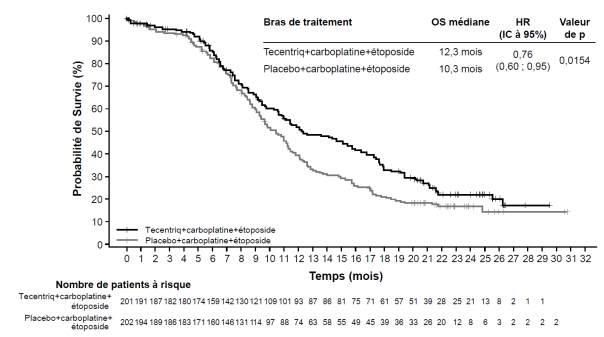
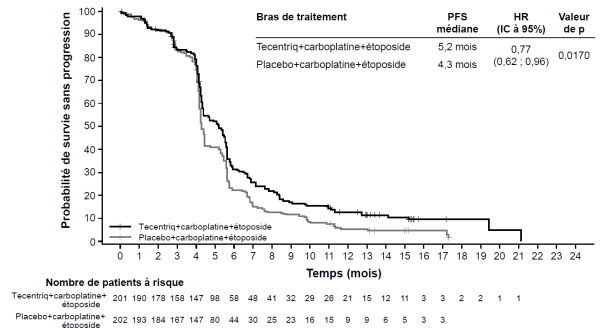
Tecentriq, en association au carboplatine et à l’étoposide, est indiqué en première ligne de traitement des patients adultes atteints d’un cancer bronchique à petites cellules (CBPC) de stade étendu (voir rubrique 5.1).
Cancer du sein triple négatif (CSTN)
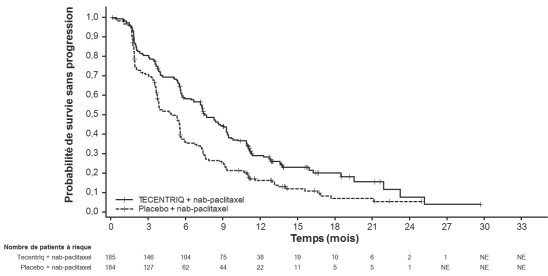
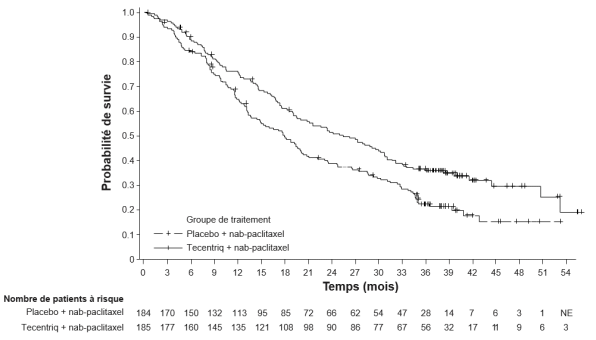
Tecentriq, en association au nab-paclitaxel, est indiqué dans le traitement des patients adultes atteints d’un cancer du sein triple négatif (CSTN) localement avancé non résécable ou métastatique, dont les tumeurs présentent une expression de PD-L1 ≥ 1 % et n’ayant pas précédemment reçu de chimiothérapie en situation métastatique.
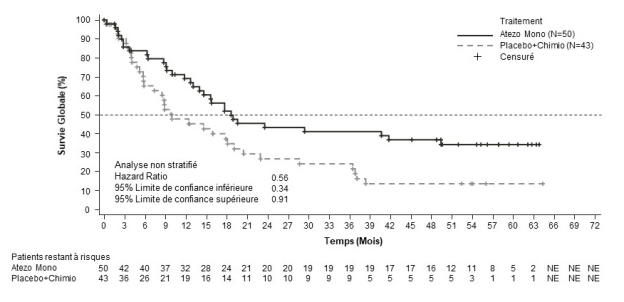
Carcinome hépatocellulaire (CHC)
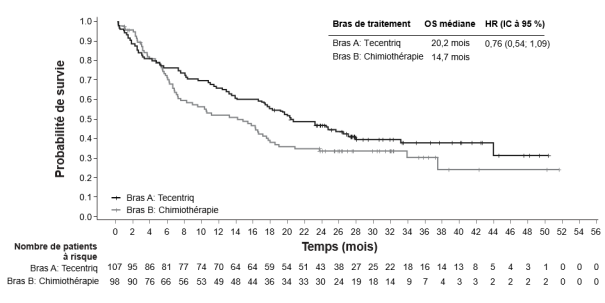
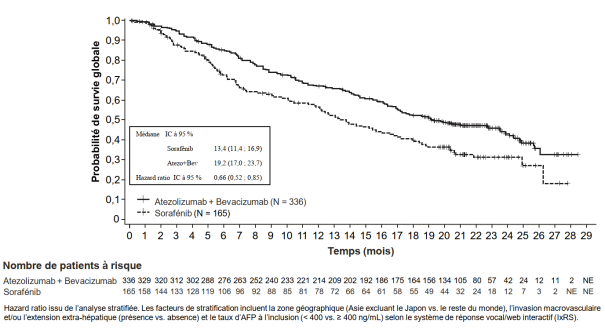
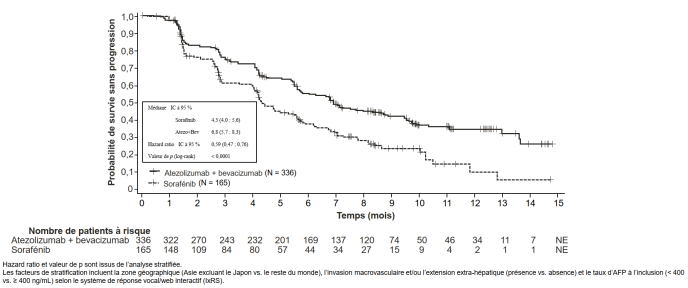
Tecentriq, en association au bevacizumab, est indiqué dans le traitement des patients adultes atteints d’un carcinome hépatocellulaire (CHC) avancé ou non résécable, n’ayant pas reçu de traitement systémique antérieur (voir rubrique 5.1).
4.2 Posologie et mode d'administration
Tecentriq doit être initié et surveillé par des médecins expérimentés dans le traitement du cancer.
Test PD-L1 pour les patients atteints d'un carcinome urothélial, d’un CSTN ou d’un CBNPC
Tecentriq en monothérapie
Si spécifiée dans l’indication, la sélection des patients à traiter par Tecentriq, sur la base de l’expression tumorale de PD-L1, doit être confirmée par un test validé (voir rubriques 4.1 et 5.1).
Tecentriq en association
Les patients atteints d'un CSTN non préalablement traité doivent être sélectionnés sur la base de l’expression tumorale de PD-L1 confirmée par un test validé (voir rubrique 5.1).
Posologie
La dose recommandée de Tecentriq est soit de 840 mg administrée par voie intraveineuse toutes les deux semaines, soit de 1 200 mg administrée par voie intraveineuse toutes les trois semaines, soit de 1 680 mg administrée par voie intraveineuse toutes les quatre semaines, comme présenté dans le Tableau 1.
Lorsque Tecentriq est administré en association, veuillez également vous référer aux RCP des produits administrés en association (voir également la rubrique 5.1).
Tableau 1 : Dose recommandée pour Tecentriq en administration par voie intraveineuse
Indication | Dose recommandée et calendrier | Durée du traitement |
Tecentriq en monothérapie |
| |
Carcinome urothélial en 1ère ligne de traitement | 840 mg toutes les 2 semaines ou | Jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable. |
CBNPC métastatique en 1ère ligne de traitement | ||
CBNPC inéligible aux sels de platine en 1ère ligne de traitement | ||
CBNPC de stade précoce | 840 mg toutes les 2 semaines ou | Pendant 1 an ou jusqu’à récidive de la maladie ou survenue d’une toxicité inacceptable. Une durée de traitement supérieure à 1 an n’a pas été étudiée. |
Carcinome urothélial en 2ème ligne de traitement | 840 mg toutes les 2 semaines ou | Jusqu’à perte du bénéfice clinique ou survenue d’une toxicité inacceptable. |
CBNPC en 2ème ligne de traitement | ||
Tecentriq en association | ||
CBNPC non-épidermoïde en 1ère ligne de traitement avec bevacizumab, paclitaxel et carboplatine | Phases d’induction et d’entretien : | Jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable. Des réponses atypiques (c’est-à-dire une progression initiale de la maladie suivie d’un rétrécissement de la tumeur) ont été observées lors de la poursuite du traitement par Tecentriq après progression de la maladie. Un traitement au-delà de la progression de la maladie peut être envisagé à la discrétion du médecin. |
CBNPC non-épidermoïde en 1ère ligne de traitement avec nab-paclitaxel et carboplatine | Phases d’induction et d’entretien : | Jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable. Des réponses atypiques (c’est-à-dire une progression initiale de la maladie suivie d’un rétrécissement de la tumeur) ont été observées lors de la poursuite du traitement par Tecentriq après progression de la maladie. Un traitement au-delà de la progression de la maladie peut être envisagé à la discrétion du médecin. |
Cancer bronchique à petites cellules (CBPC) de stade étendu en 1ère ligne de traitement avec carboplatine et étoposide | Phases d’induction et d’entretien : | Jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable. Des réponses atypiques (c’est-à-dire une progression initiale de la maladie suivie d’un rétrécissement de la tumeur) ont été observées lors de la poursuite du traitement par Tecentriq après progression de la maladie. Un traitement au-delà de la progression de la maladie peut être envisagé à la discrétion du médecin. |
Cancer du sein triple négatif (CSTN) localement avancé non résécable ou métastatique en 1ère ligne de traitement avec nab-paclitaxel | 840 mg toutes les 2 semaines ou | Jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable. |
Carcinome hépatocellulaire (CHC) avancé ou non résécable avec bevacizumab | 840 mg toutes les 2 semaines ou | Jusqu’à perte du bénéfice clinique ou survenue d’une toxicité inacceptable. |
Oubli ou retard de dose
Si une dose programmée de Tecentriq est oubliée, elle doit être administrée dès que possible. Le calendrier d'administration devra être modifié de manière à conserver l’intervalle approprié entre les doses.
Modifications de dose pendant le traitement
Les réductions de dose de Tecentriq ne sont pas recommandées.
Retard de dose ou arrêt d'administration (voir également les rubriques 4.4 et 4.8)
Tableau 2 : Recommandations de modification de dose pour Tecentriq
Effet indésirable à médiation immunitaire | Sévérité | Modification du traitement |
Pneumopathie inflammatoire | Grade 2 | Suspendre Tecentriq. |
| Grade 3 ou 4 | Arrêt définitif de Tecentriq. |
Hépatite chez les patients non atteints d’un carcinome hépatocellulaire (CHC) | Grade 2 : | Suspendre Tecentriq. |
| Grade 3 ou 4 : | Arrêt définitif de Tecentriq. |
Hépatite chez les patients atteints d’un CHC | Si ALAT ou ASAT dans les limites normales à l’initiation et augmentation > 3 x à ≤ 10 x LSN | Suspendre Tecentriq. |
Si augmentation de l’ALAT ou ASAT > 10 x LSN | Arrêt définitif de Tecentriq. | |
Colite | Diarrhée de grade 2 ou 3 (augmentation ≥ 4 selles/jour depuis le début du traitement) | Suspendre Tecentriq. |
| Diarrhée de grade 4 ou colite de grade 4 (mettant en jeu le pronostic vital ; intervention urgente requise) | Arrêt définitif de Tecentriq. |
Hypothyroïdie ou hyperthyroïdie | Symptomatique | Suspendre Tecentriq. |
Insuffisance surrénalienne | Symptomatique | Suspendre Tecentriq. |
Hypophysite | Grade 2 ou 3 | Suspendre Tecentriq. |
Grade 4 | Arrêt définitif de Tecentriq. | |
Diabète de type 1 | Hyperglycémie de grade 3 ou 4 (glucose à jeun > 250 mg/dL ou 13,9 mmol/L) | Suspendre Tecentriq. |
Éruption cutanée/réactions cutanées sévères | Grade 3 | Suspendre Tecentriq. |
| Grade 4 | Arrêt définitif de Tecentriq. |
Syndrome myasthénique/ | Parésie faciale de grade 1 ou 2 | Suspendre Tecentriq. |
Tous grades de syndrome myasthénique/myasthénie, syndrome de Guillain-Barré et méningo-encéphalite | Arrêt définitif de Tecentriq. | |
Myélite | Grade 2, 3 ou 4 | Arrêt définitif de Tecentriq. |
Pancréatite | Augmentation des taux sériques d'amylase ou de lipase de grade 3 ou 4 | Suspendre Tecentriq. |
Pancréatite de grade 4 ou récidive de pancréatite, quel que soit le grade | Arrêt définitif de Tecentriq. | |
Myocardite | Grade 2 ou supérieur | Arrêt définitif de Tecentriq. |
Néphrite | Grade 2 | Suspendre Tecentriq. |
Grade 3 ou 4 | Arrêt définitif de Tecentriq. | |
Myosite | Grade 2 ou 3 | Suspendre Tecentriq. |
Myosite de grade 4 ou de grade 3 récurrente | Arrêt définitif de Tecentriq. | |
Troubles péricardiques | Péricardite de grade 1 | Suspendre Tecentriq2. |
Grade 2 ou supérieur | Arrêt définitif de Tecentriq. | |
Lymphohistiocytose hémophagocytaire | Suspicion de lymphohistiocytose hémophagocytaire1 | Arrêt définitif de Tecentriq. |
Autres effets indésirables à médiation immunitaire | Grade 2 ou 3 | Suspendre Tecentriq jusqu’à ce que les effets indésirables s’améliorent jusqu’au grade 0 ou au grade 1 dans les 12 semaines et que la dose de corticoïdes a été réduite à ≤ 10 mg de prednisone ou équivalent par jour. |
Grade 4 ou grade 3 récurrent | Arrêt définitif de Tecentriq | |
Autres effets indésirables | Sévérité | Modification du traitement |
Réactions liées à la perfusion | Grade 1 ou 2 | Réduire le débit de perfusion ou interrompre la perfusion. |
Grade 3 ou 4 | Arrêt définitif de Tecentriq. |
ALAT : alanine aminotransférase ; ASAT : aspartate aminotransférase ; LSN : limite supérieure de la normale
Remarque : la toxicité doit être évaluée selon la version actuelle du National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE).
1 Quelle que soit la sévérité
2 Effectuer une évaluation cardiaque détaillée pour déterminer l'étiologie et prendre en charge de manière appropriée
Populations particulières
Population pédiatrique
La sécurité et l'efficacité de Tecentriq chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies. Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Personnes âgées
Sur la base d’une analyse pharmacocinétique de population, aucune adaptation posologique de Tecentriq n'est requise chez les patients âgés de 65 ans et plus (voir rubriques 4.8 et 5.1).
Patients asiatiques
En raison d’une augmentation des toxicités hématologiques observée chez les patients asiatiques au cours de l’essai clinique IMpower150, il est recommandé que la dose initiale de paclitaxel soit de 175 mg/m2 toutes les trois semaines.
Insuffisance rénale
Sur la base d’une analyse pharmacocinétique de population, aucune adaptation posologique n'est requise chez les patients atteints d’insuffisance rénale légère ou modérée (voir rubrique 5.2). Les données chez les patients atteints d’insuffisance rénale sévère sont trop limitées pour tirer des conclusions dans cette population.
Insuffisance hépatique
Sur la base d’une analyse pharmacocinétique de population, aucune adaptation posologique n'est requise chez les patients atteints d'insuffisance hépatique légère ou modérée. Tecentriq n’a pas été étudié chez les patients atteints d'insuffisance hépatique sévère (voir rubrique 5.2).
Indice de performance Eastern Cooperative Oncology Group (ECOG) ≥ 2
Les patients avec un indice de performance ECOG ≥ 2 étaient exclus des essais cliniques dans le CSTN, le CBPC de stade étendu, en deuxième ligne du carcinome urothélial et dans le CHC (voir rubriques 4.4 et 5.1).
Mode d'administration
Il est important de vérifier les étiquettes du produit pour s’assurer que la formulation correcte (intraveineuse ou sous-cutanée) est administrée au patient, conformément à la prescription.
La formulation intraveineuse de Tecentriq n’est pas destinée à l’administration sous-cutanée et doit être administrée uniquement par perfusion intraveineuse. Les perfusions ne doivent pas être administrées en injection rapide ou bolus intraveineux.
Les patients recevant actuellement Tecentriq par voie intraveineuse peuvent passer à la solution injectable par voie sous-cutanée d'atezolizumab ou vice versa.
La dose initiale de Tecentriq par voie intraveineuse doit être administrée en 60 minutes. Si la première perfusion est bien tolérée, toutes les perfusions suivantes peuvent être administrées en 30 minutes.
Pour les instructions concernant la dilution et la manipulation du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à l'atezolizumab ou à l'un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
La sécurité d’atezolizumab en monothérapie est basée sur les données groupées de 5 039 patients atteints de différents types de tumeurs. Les effets indésirables les plus fréquents (> 10 %) étaient les suivants : fatigue (29,3 %), diminution de l'appétit (20,1 %), éruptions cutanées (19,7 %), nausées (18,8 %), toux (18,2 %), diarrhée (18,1 %), fièvre (17,9 %), dyspnée (16,6 %), arthralgie (16,2 %), prurit (13,3 %), asthénie (13 %), dorsalgie (12,2 %), vomissements (11,7 %), infection des voies urinaires (11 %) et céphalées (10,2 %).
La sécurité d’atezolizumab administré en association à d’autres médicaments, a été évaluée chez 4 535 patients atteints de différents types de tumeurs. Les effets indésirables les plus fréquents (≥ 20 %) étaient les suivants : anémie (36,8 %), neutropénie (36,6 %), nausées (35,5 %), fatigue (33,1 %), alopécie (28,1 %), éruption cutanée (27,8 %), diarrhées (27,6 %), thrombopénie (27,1 %), constipation (25,8 %), diminution de l’appétit (24,7 %) et neuropathie périphérique (24,4 %).
Utilisation d’atezolizumab dans le traitement adjuvant d’un CBNPC
Le profil de sécurité d’atezolizumab en traitement adjuvant dans la population de patients atteints d’un cancer bronchique non à petites cellules (CBNPC) (IMpower010) était généralement cohérent avec le profil de sécurité global en monothérapie chez les patients atteints d’un cancer avancé. Cependant, l’incidence des effets indésirables à médiation immunitaire d’atezolizumab dans l’essai IMpower010 a été de 51,7 %, comparée à 38,4 % dans la population globale en monothérapie chez les patients avec un cancer avancé. Aucun nouvel effet indésirable à médiation immunitaire n’a été identifié en traitement adjuvant.
Utilisation d’atezolizumab en association au bevacizumab, paclitaxel et carboplatine
Dans l'essai clinique dans le CBNPC en première ligne (IMpower150), une fréquence globale plus élevée d'effets indésirables a été observée dans le schéma thérapeutique avec les quatre médicaments atezolizumab, bevacizumab, paclitaxel et carboplatine, comparativement à atezolizumab, paclitaxel et carboplatine, dont des effets de grade 3 et 4 (63,6 % comparé à 57,5 %), des effets de grade 5 (6,1 % comparé à 2,5 %), des effets indésirables d’intérêt particulier pour l’atezolizumab (52,4 % comparé à 48,0 %), ainsi que des effets indésirables conduisant à un arrêt de l’un des traitements (33,8 % comparé à 13,3 %). Nausée, diarrhée, stomatite, fatigue, fièvre, inflammation des muqueuses, diminution de l'appétit, perte de poids, hypertension et protéinurie ont été plus fréquemment rapportées (différence ≥ 5 %) chez les patients recevant l'atezolizumab en association au bevacizumab, paclitaxel et carboplatine. Les autres effets indésirables cliniquement significatifs qui ont été observés plus fréquemment dans le bras atezolizumab, bevacizumab, paclitaxel et carboplatine étaient les suivants : épistaxis, hémoptysie, accident vasculaire cérébral, dont des événements d’évolution fatale.
Des informations complémentaires sur les effets indésirables graves sont présentées à la rubrique 4.4.
Tableau des effets indésirables
Les effets indésirables sont listés selon la classification MedDRA par organe et par fréquence dans le Tableau 3 pour l’atezolizumab administré en monothérapie ou en association. Les effets indésirables connus pour l’atezolizumab ou des chimiothérapies administrées seules peuvent survenir durant le traitement avec ces médicaments en association, même si ces effets n’ont pas été rapportés dans les essais cliniques avec des traitements associés. Les catégories suivantes de fréquence ont été utilisées : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 : Résumé des effets indésirables survenant chez les patients traités par l’atezolizumab
Atezolizumab en monothérapie | Atezolizumab en association | |
Infections et infestations | ||
Très fréquent | infection des voies urinairesa | infection pulmonaireb |
Fréquent |
| sepsisaj |
Rare | infection à cytomégalovirus | infection à cytomégalovirus |
Affections hématologiques et du système lymphatique | ||
Très fréquent |
| anémie, thrombopénied, neutropéniee, leucopénief |
Fréquent | thrombopénied, neutropéniee | lymphopénieg |
Rare | lymphohistiocytose hémophagocytaire, anémie hémolitique autoimmuneav | lymphohistiocytose hémophagocytaire, anémie hémolitique autoimmuneav |
Affections du système immunitaire | ||
Fréquent | réaction liée à la perfusionh | réaction liée à la perfusionh |
Rare | sarcoïdosear |
|
Affections endocriniennes | ||
Très fréquent |
| hypothyroïdiei |
Fréquent | hypothyroïdiei, hyperthyroïdiej | hyperthyroïdiej |
Peu fréquent | diabètek, insuffisance surrénaliennel, hypophysitem | hypophysitem |
Troubles du métabolisme et de la nutrition | ||
Très fréquent | diminution de l’appétit | diminution de l’appétit |
Fréquent | hypokaliémieae, hyponatrémieaf, hyperglycémie, hypoalbuminémie, hypophosphatémie, hypocalcémie | hypokaliémieae, hyponatrémieaf, hypomagnésémien, hypoalbuminémie, hypophosphatémie, hypocalcémie |
Affections du système nerveux | ||
Très fréquent | céphalée | neuropathie périphériqueo, céphalée |
Fréquent | neuropathie périphériqueo | syncope, vertige |
Peu fréquent | syndrome de Guillain-Barrép, méningo-encéphaliteq |
|
Rare | syndrome myasthéniquer, parésie faciale, myélite | parésie faciale |
Affections oculaires | ||
Peu fréquent | uvéiteas |
|
Rare |
| uvéiteas |
Affections cardiaques | ||
Fréquent | troubles péricardiquesao |
|
Peu fréquent |
| troubles péricardiquesao |
Rare | myocardites |
|
Affections vasculaires | ||
Très fréquent |
| hypertensionai |
Fréquent | hypotension |
|
Affections respiratoires, thoraciques et médiastinales | ||
Très fréquent | dyspnée, toux | dyspnée, toux, rhinopharyngiteam |
Fréquent | pneumopathie inflammatoiret, hypoxieag, rhinopharyngiteam | dysphonie |
Affections gastro-intestinales | ||
Très fréquent | nausées, vomissements, diarrhéesu | nausées, vomissements, diarrhéesu, constipation |
Fréquent | colitev, douleur abdominale, dysphagie, douleur oropharyngéew, sécheresse buccale | stomatite, dysgueusie, colitev |
Peu fréquent | pancréatitex |
|
Rare | maladie cœliaque | maladie cœliaque |
Affections hépatobiliaires | ||
Fréquent | augmentation du taux d’ASAT, augmentation du taux d’ALAT, hépatitey | augmentation du taux d’ASAT, augmentation du taux d’ALAT |
Affections de la peau et du tissu sous-cutané | ||
Très fréquent | éruption cutanéez, prurit | éruption cutanéez, prurit, alopécieah |
Fréquent | sécheresse cutanéeap |
|
Peu fréquent | réactions cutanées sévèresak, psoriasisan, lichenaq | réactions cutanées sévèresak, psoriasisan |
Rare | pemphigoïde | pemphigoïde, lichenaq |
Affections musculo-squelettiques et systémiques | ||
Très fréquent | arthralgie, dorsalgie | arthralgie, douleur musculo-squelettiqueaa, dorsalgie |
Fréquent | douleur musculo-squelettiqueaa, arthriteat | arthriteat |
Peu fréquent | myositeab, ténosynoviteau | ténosynoviteau |
Affections du rein et des voies urinaires | ||
Fréquent | augmentation de la créatininémiec | protéinurieac, augmentation de la créatininémiec |
Peu fréquent | néphritead |
|
Fréquence indéterminée | cystite non infectieuseal |
|
Troubles généraux et anomalies au site d’administration | ||
Très fréquent | fièvre, fatigue, asthénie | fièvre, fatigue, asthénie, oedème périphérique |
Fréquent | syndrome pseudo-grippal, frissons |
|
Investigations | ||
Fréquent | augmentation du taux de gamma-glutamyl transférase | augmentation du taux de phosphatase alcaline dans le sang, augmentation du taux de gamma-glutamyl transférase |
Peu fréquent | augmentation de la créatine phosphokinase sanguine |
|
a Inclut des cas rapportés d’infection des voies urinaires, de cystite, de pyélonéphrite, d’infection des voies urinaires par colibacille, d’infection des voies urinaires bactérienne, d’infection rénale, de pyélonéphrite aiguë, de pyélonéphrite chronique, de pyélite, d’abcès rénal, d’infection streptococcique des voies urinaires, d’urétrite, d’infection des voies urinaires fongique, d’infection des voies urinaires à pseudomonas.
b Inclut des cas rapportés de pneumonie, de bronchite, d’infection des voies respiratoires basses, d’épanchement pleural infectieux, de trachéobronchite, de pneumonie atypique, d’abcès pulmonaire, d’exacerbation infectieuse d’une affection chronique obstructive des voies aériennes, de pneumopathie paranéoplasique, de pyopneumothorax, de pleurésie, de pneumonie post-intervention.
c Inclut des cas rapportés d’augmentation de la créatininémie, d’hypercréatininémie.
d Inclut des cas rapportés de thrombopénie immunitaire (issus d’études non incluses dans les données groupées), de thrombopénie, de diminution du nombre de plaquettes.
e Inclut des cas rapportés de neutropénie, de diminution du nombre de neutrophiles, de neutropénie fébrile, de sepsis neutropénique, de granulocytopénie.
f Inclut des cas rapportés de diminution du nombre de globules blancs, de leucopénie.
g Inclut des cas rapportés de lymphopénie, de diminution du nombre de lymphocytes.
h Inclut des cas rapportés de réaction liée à la perfusion, de syndrome de relargage des cytokines, d’hypersensibilité, d’anaphylaxie.
i Inclut des cas rapportés de positivité aux anticorps anti-thyroïdiens, d’hypothyroïdie auto-immune, de thyroïdite auto-immune, de diminution de la thyréostimuline (TSH), d'augmentation de la thyréostimuline (TSH), de syndrome euthyroïdien, de goitre, d'hypothyroïdie, d’hypothyroïdie à médiation immunitaire, de thyroïdite à médiation immunitaire, de myxœdème, d’hypothyroïdie primaire, de troubles thyroïdiens, de diminution des hormones thyroïdiennes, de paramètres fonctionnels thyroïdiens anormaux, de thyroïdite, de thyroïdite aiguë, de diminution de la thyroxine, de diminution de la thyroxine libre, d’augmentation de la thyroxine libre, d’augmentation de la thyroxine, de diminution de la tri-iodothyronine, d’augmentation de la tri-iodothyronine, de tri-iodothyronine libre anormale, de diminution de la tri-iodothyronine libre, d’augmentation de la tri-iodothyronine libre, de thyroïdite silencieuse.
j Inclut des cas rapportés d'hyperthyroïdie, de maladie de Basedow, d’ophtalmopathie endocrinienne, d’exophtalmie.
k Inclut des cas rapportés de diabète, de diabète de type 1, d’acidocétose diabétique, d’acidocétose.
l Inclut des cas rapportés d’insuffisance surrénalienne, de diminution de la corticotropine sanguine, de déficit en glucocorticoïdes, d’insuffisance surrénalienne primaire, d’insuffisance adrénocorticale secondaire.
m Inclut des cas rapportés d’hypophysite, d’hypopituitarisme, d’insuffisance adrénocorticale secondaire, de trouble de la régulation de la température.
n Inclut des cas rapportés d’hypomagnésémie, de diminution du magnésium dans le sang.
o Inclut des cas rapportés de neuropathie périphérique, de neuropathie auto-immune, de neuropathie sensitive périphérique, de polyneuropathie, de zona, de neuropathie motrice périphérique, d’amyotrophie névralgique, de neuropathie sensitivo motrice périphérique, de neuropathie toxique, de neuropathie axonale, de plexopathie lombo-sacrée, d’arthropathie neuropathique, d’infection des nerfs périphériques, de névrite, de neuropathie à médiation immunitaire.
p Inclut des cas rapportés de syndrome de Guillain-Barré, de paralysie flasque ascendante, de polyneuropathie démyélinisante.
q Inclut des cas rapportés d'encéphalite, d'encéphalite auto-immune, de méningite, de méningite aseptique, de photophobie.
r Inclut des cas rapportés de myasthénie grave.
s Inclut des cas rapportés de myocardite, de myocardite auto-immune et de myocardite à médiation immunitaire.
t Inclut des cas rapportés de pneumopathie inflammatoire, d'infiltration pulmonaire, de bronchiolite, de pneumopathie à médiation immunitaire, de pneumopathie inflammatoire à médiation immunitaire, de pneumopathie interstitielle, d’alvéolite, d’opacité pulmonaire, de fibrose pulmonaire, de toxicité pulmonaire, de pneumopathie radique.
u Inclut des cas rapportés de diarrhée, de selles impérieuses, de selles fréquentes, d’hypermotilité gastro-intestinale.
v Inclut des cas rapportés de colite, de colite auto-immune, de colite ischémique, de colite microscopique, de colite ulcéreuse, de colite de dérivation, de colite à éosinophiles, d’entérocolite à médiation immunitaire.
w Inclut des cas rapportés de douleur oropharyngée, d’inconfort oropharyngé, d’irritation de la gorge.
x Inclut des cas rapportés de pancréatite auto-immune, de pancréatite, de pancréatite aiguë, de lipase augmentée, d’amylase augmentée.
y Inclut des cas rapportés d’ascite, d'hépatite auto-immune, de cytolyse hépatique, d'hépatite, d’hépatite aiguë, d’hépatite toxique, d’hépatotoxicité, d’hépatite à médiation immunitaire, de trouble hépatique, de lésion hépatique d’origine médicamenteuse, d’insuffisance hépatique, de stéatose hépatique, de lésion hépatique, d’atteintes hépatiques, d’hémorragie de varices œsophagiennes, de varices œsophagiennes, de péritonite bactérienne spontanée.
z Inclut des cas rapportés d’acné, de bulle, de dermatite, de dermatite acnéiforme, de dermatite allergique, d’éruption médicamenteuse, d’eczéma, d’eczéma infecté, d’érythème, d’érythème de la paupière, d’éruption cutanée de la paupière, d’érythème pigmenté fixe, de folliculite, de furoncle, de dermatite des mains, de dermatite à médiation immunitaire, de bulle labiale, de bulle hémorragique buccale, de syndrome d’érythrodysesthésie palmo-plantaire, de pemphigoïde, d’éruption cutanée, d’éruption cutanée érythémateuse, d’éruption cutanée maculaire, d’éruption cutanée maculo-papuleuse, d’éruption cutanée morbilliforme, d’éruption cutanée papuleuse, d’éruption cutanée papulosquameuse, d’éruption cutanée pruritique, d’éruption cutanée pustuleuse, d’éruption cutanée vésiculaire, de dermatite scrotale, de dermatite séborrhéique, d’exfoliation cutanée, de toxicité cutanée, d’ulcère cutané, d’éruption cutanée du site d’abord vasculaire.
aa Inclut des cas rapportés de douleur musculo-squelettique, de myalgie, de douleur osseuse.
ab Inclut des cas rapportés de myosite, de rhabdomyolyse, de pseudopolyarthrite rhizomélique, de dermatomyosite, d’abcès musculaire, de myoglobinurie, de myopathie, de polymyosite.
ac Inclut des cas rapportés de protéinurie, de présence de protéines dans les urines, d’hémoglobinurie, d’anomalie des urines, de syndrome néphrotique, d’albuminurie.
ad Inclut des cas rapportés de néphrite, de néphrite auto-immune, de néphropathie du purpura rhumatoïde (purpura de Henoch-Schönlein), de glomérulonéphrite paranéoplasique, de néphrite tubulo-interstitielle.
ae Inclut des cas rapportés d’hypokaliémie, de diminution du potassium dans le sang.
af Inclut des cas rapportés d’hyponatrémie, de diminution du sodium dans le sang.
ag Inclut des cas rapportés d’hypoxie, de diminution de la saturation en oxygène, de pO2 diminuée.
ah Inclut des cas rapportés d’alopécie, de madarose, d’alopécie en plaque, d’alopécie totale, d’hypotrichose.
ai Inclut des cas rapportés d’hypertension, d’augmentation de la pression artérielle, de crise hypertensive, d’augmentation de la pression artérielle systolique, d’hypertension diastolique, de pression artérielle insuffisamment contrôlée, de rétinopathie hypertensive, de néphropathie hypertensive, d’hypertension essentielle, d’hypertension orthostatique.
aj Inclut des cas rapportés de sepsis, de choc septique, d’urosepsis, de sepsis neutropénique, de sepsis pulmonaire, de sepsis bactérien, de sepsis à klebsiella, de sepsis abdominal, de sepsis à candida, de sepsis à escherichia, de sepsis à pseudomonas, de sepsis à staphylocoques.
ak Inclut des cas rapportés de dermatite bulleuse, d’éruption cutanée exfoliative, d’érythème polymorphe, de dermatite exfoliative, de dermatite exfoliative généralisée, de toxidermie, de syndrome de Stevens-Johnson, de réaction médicamenteuse avec éosinophilie et symptômes systémiques, de nécrolyse épidermique toxique, de vascularite cutanée.
al Inclut des cas rapportés de cystite non infectieuse et de cystite à médiation immunitaire.
am Inclut des cas rapportés de rhinopharyngite, de congestion nasale, de rhinorrhée.
an Inclut des cas rapportés de psoriasis, de dermatite psoriasiforme.
ao Inclut des cas rapportés de péricardites, d’épanchement péricardique, de tamponnade cardiaque et de péricardite constrictive.
ap Inclut des cas rapportés de sécheresse cutanée, de xérose.
aq Inclut des cas rapportés de kératose lichénoïde, de lichen scléreux et de lichen plan.
ar Inclut des cas rapportés de sarcoïdose, de sarcoïdose pulmonaire et de sarcoïdose de ganglion lymphatique.
as Inclut des cas rapportés d’uvéite, d’iridocyclite et d’iritis.
at Inclut des cas rapportés d’arthrite, de gonflement des articulations, d’arthrose, de polyarthrite rhumatoïde, de polyarthrite, d’arthrose rachidienne, d’arthrite auto-immune, d’arthrite à médiation immunitaire, de spondylite, d’épanchement articulaire, d’arthropathie, d’oligoarthrite, d’affection rhumatismale.
au Inclut des cas rapportés de tendinite, de douleur aux tendons, de ténosynovite et de synovite.
av Inclut des cas rapportés d’anémie hémolitique autoimmune, d’anémie hémolitique.
Description d’effets indésirables sélectionnés
Les données ci-dessous reflètent les informations concernant les effets indésirables significatifs observés avec l’atezolizumab en monothérapie dans les essais cliniques (voir rubrique 5.1). Des informations complémentaires sur les effets indésirables significatifs observés avec l’atezolizumab administré en association sont présentées en cas de différences cliniquement pertinentes par rapport à l’atezolizumab en monothérapie. Les recommandations de prise en charge de ces effets indésirables sont décrites aux rubriques 4.2 et 4.4.
Pneumopathie inflammatoire à médiation immunitaire
Une pneumopathie inflammatoire est survenue chez 3,0 % (151/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Parmi ces patients, trois ont présenté des événements d’évolution fatale. Le délai médian de survenue a été de 3,7 mois (intervalle de 3 jours à 29,8 mois). La durée médiane de l’événement indésirable a été de 1,7 mois (intervalle de 0 jour à 27,8+ mois ; + signale une valeur censurée). La pneumopathie a conduit à l'arrêt d'atezolizumab chez 41 patients (0,8 %). Une pneumopathie inflammatoire nécessitant l’utilisation de corticoïdes est survenue chez 1,8 % (92/5 039) des patients recevant l’atezolizumab en monothérapie.
Hépatite à médiation immunitaire
Une hépatite est survenue chez 1,7 % (88/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Parmi ces 88 patients, trois ont présenté des événements d’évolution fatale. Le délai médian de survenue a été de 1,4 mois (intervalle de 0 jour à 26,3 mois). La durée médiane de l’événement indésirable a été de 1 mois (intervalle de 0 jour à 52,1+ mois ; + signale une valeur censurée). L’hépatite a conduit à l’arrêt d’atezolizumab chez 46 (0,9 %) patients. Une hépatite nécessitant l’utilisation de corticoïdes est survenue chez 2,6 % (130/5 039) des patients recevant l’atezolizumab en monothérapie.
Colite à médiation immunitaire
Une colite est survenue chez 1,2 % (62/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 4,5 mois (intervalle de 15 jours à 36,4 mois). La durée médiane de l’événement indésirable a été de 1,4 mois (intervalle de 3 jours à 50,2+ mois ; + signale une valeur censurée). La colite a conduit à l'arrêt d'atezolizumab chez 24 (0,5 %) patients. Une colite nécessitant l’utilisation de corticoïdes est survenue chez 0,6 % (30/5 039) des patients recevant l’atezolizumab en monothérapie.
Endocrinopathies à médiation immunitaire
Troubles de la thyroïde
Une hypothyroïdie est survenue chez 8,5 % (427/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 4,2 mois (intervalle de 0 jour à 38,5 mois). Une hypothyroïdie est survenue chez 17,4 % (86/495) des patients ayant reçu l’atezolizumab en monothérapie en traitement adjuvant d’un CBNPC. Le délai médian de survenue a été de 4,0 mois (intervalle de 22 jours à 11,8 mois).
Une hyperthyroïdie est survenue chez 2,4 % (121/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 2,7 mois (intervalle de 0 jour à 24,3 mois). Une hyperthyroïdie est survenue chez 6,5 % (32/495) des patients ayant reçu l’atezolizumab en monothérapie en traitement adjuvant d’un CBNPC. Le délai médian de survenue a été de 2,8 mois (intervalle de 1 jour à 9,9 mois).
Insuffisance surrénalienne
Une insuffisance surrénalienne est survenue chez 0,5 % (25/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 6,2 mois (intervalle de 3 jours à 21,4 mois). Une insuffisance surrénalienne a conduit à l’arrêt d'atezolizumab chez 5 (0,1 %) patients. Une insuffisance surrénalienne nécessitant l’utilisation de corticoïdes est survenue chez 0,4 % (20/5 039) des patients recevant l’atezolizumab en monothérapie.
Hypophysite
Une hypophysite est survenue chez 0,2 % (9/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 5,3 mois (intervalle de 21 jours à 13,7 mois). Chez six patients (0,1 %), l’hypophysite a nécessité l’utilisation de corticoïdes et le traitement par l’atezolizumab a été arrêté chez un patient (< 0,1 %).
Une hypophysite est survenue chez 1,4 % (15/1 093) des patients ayant reçu l’atezolizumab en association au paclitaxel suivi de l’atezolizumab, de la doxorubicine ou de l’épirubicine à dose-densité, et de la cyclophosphamide. Le délai médian de survenue a été de 3,8 mois (intervalle de 2,4 à 10,7 mois). Chez onze patients (1,0 %), l’hypophysite a nécessité l’utilisation de corticoïdes. Le traitement par l’atezolizumab a été arrêté chez sept patients (0,6 %).
Une hypophysite est survenue chez 0,8 % (3/393) des patients ayant reçu l’atezolizumab avec le bevacizumab, le paclitaxel et le carboplatine. Le délai médian de survenue a été de 7,7 mois (intervalle de 5,0 à 8,8 mois). Chez deux patients, l’hypophysite a nécessité l’utilisation de corticoïdes.
Une hypophysite est survenue chez 0,4 % (2/473) des patients ayant reçu l’atezolizumab en association au nab-paclitaxel et au carboplatine. Le délai médian de survenue a été de 5,2 mois (intervalle de 5,1 à 5,3 mois). Chez les deux patients, l’hypophysite a nécessité l’utilisation de corticoïdes.
Diabète
Un diabète est survenu chez 0,6 % (30/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 5,5 mois (intervalle de 3 jours à 29,0 mois). Le diabète a conduit à l'arrêt d’atezolizumab chez < 0,1 % (3/5 039) des patients. Quatre (< 0,1 %) patients ont nécessité l’utilisation de corticoïdes.
Un diabète est survenu chez 2,0 % (10/493) des patients atteints d’un CHC ayant reçu l’atezolizumab en association au bevacizumab. Le délai médian de survenue a été de 4,4 mois (intervalle de 1,2 mois à 8,3 mois). Aucun cas de diabète n’a conduit à l'arrêt d’atezolizumab.
Méningo-encéphalite à médiation immunitaire
Une méningo-encéphalite est survenue chez 0,4 % (22/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 15 jours (intervalle de 0 jour à 12,5 mois). La durée médiane de l’événement indésirable a été de 24 jours (intervalle de 6 jours à 14,5+ mois ; + signale une valeur censurée).
Une méningo-encéphalite nécessitant l’utilisation de corticoïdes est survenue chez 0,2 % (12/5 039) des patients recevant l’atezolizumab et huit patients (0,2 %) ont arrêté l’atezolizumab.
Neuropathies à médiation immunitaire
Syndrome de Guillain-Barré et polyneuropathie démyélinisante
Un syndrome de Guillain-Barré et une polyneuropathie démyélinisante sont survenus chez 0,1 % (6/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 4,1 mois (intervalle de 18 jours à 8,1 mois). La durée médiane de l’événement indésirable a été de 8,0 mois (intervalle de 18 jours à 24,5+ mois ; + signale une valeur censurée). Le syndrome de Guillain-Barré a conduit à l’arrêt d’atezolizumab chez 1 patient (< 0,1 %). Un syndrome de Guillain-Barré nécessitant l’utilisation de corticoïdes est survenu chez < 0,1 % (3/5 039) des patients recevant l’atezolizumab en monothérapie.
Parésie faciale à médiation immunitaire
Une parésie faciale est survenue chez < 0,1 % (1/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai de survenue a été de 29 jours. La durée a été de 1,1 mois. L’événement n’a pas nécessité l’utilisation de corticoïdes et n’a pas conduit à l’arrêt d’atezolizumab.
Myélite à médiation immunitaire
Une myélite est survenue chez < 0,1 % (1/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai de survenue a été de 3 jours. L’événement a nécessité l’utilisation de corticoïdes mais n’a pas conduit à l’arrêt d’atezolizumab.
Syndrome myasthénique
Une myasthénie est survenue chez < 0,1 % (2/5 039) des patients (incluant un cas de décès) ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 2,6 mois (intervalle de 1,2 mois à 4 mois).
Pancréatite à médiation immunitaire
Une pancréatite, incluant une augmentation de l'amylase et de la lipase, est survenue chez 0,8 % (40/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 5 mois (intervalle de 0 jour à 24,8 mois). La durée médiane de l’événement indésirable a été de 24 jours (intervalle de 3 jours à 40,4+ mois ; + signale une valeur censurée). La pancréatite a conduit à l’arrêt d’atezolizumab chez 3 (< 0,1 %) patients. Une pancréatite nécessitant l’utilisation de corticoïdes est survenue chez 0,2 % (8/5 039) des patients recevant l’atezolizumab en monothérapie.
Myocardite à médiation immunitaire
Une myocardite est survenue chez < 0,1 % (5/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Sur les 5 patients, un a présenté un événement d’évolution fatale en traitement adjuvant d’un CBNPC. Le délai médian de survenue a été de 3,7 mois (intervalle de 1,5 à 4,9 mois). La durée médiane de l’événement indésirable a été de 14 jours (intervalle de 12 jours à 2,8 mois). La myocardite a conduit à l’arrêt d’atezolizumab chez 3 (< 0,1 %) patients. Trois (< 0,1 %) patients ont nécessité l’utilisation de corticoïdes.
Néphrite à médiation immunitaire
Une néphrite est survenue chez 0,2 % (11/5 039) des patients ayant reçu l’atezolizumab. Le délai médian de survenue était de 5,1 mois (intervalle de 3 jours à 17,5 mois). Une néphrite a conduit à l’arrêt d’atezolizumab chez 5 (≤ 0,1 %) patients. Cinq (0,1 %) patients ont nécessité l’utilisation de corticoïdes.
Myosite à médiation immunitaire
Une myosite est survenue chez 0,6 % (32/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 3,5 mois (intervalle de 12 jours à 11,5 mois). La durée médiane était de 3,2 mois (intervalle de 9 jours à 51,1+ mois ; + signale une valeur censurée). La myosite a conduit à l’arrêt d’atezolizumab chez 6 patients (0,1 %). Dix (0,2 %) patients ont nécessité l’utilisation de corticoïdes.
Réactions cutanées sévères à médiation immunitaire
Des réactions cutanées sévères sont survenues chez 0,6 % (30/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Sur ces 30 patients, un a présenté un événement d’évolution fatale. Le délai médian de survenue a été de 4,8 mois (intervalle de 3 jours à 15,5 mois). La durée médiane était de 2,4 mois (intervalle de 1 jour à 37,5+ mois ; + signale une valeur censurée). Les réactions cutanées sévères ont conduit à l’arrêt d’atezolizumab chez 3 (< 0,1 %) patients. Des réactions cutanées sévères nécessitant l’utilisation de corticoïdes systémiques sont survenues chez 0,2 % (9/5 039) des patients recevant l’atezolizumab en monothérapie.
Troubles péricardiques à médiation immunitaire
Des troubles péricardiques sont survenus chez 1 % (49/5 039) des patients ayant reçu l’atezolizumab en monothérapie. Le délai médian de survenue a été de 1,4 mois (intervalle de 6 jours à 17,5 mois). La durée médiane était de 2,5 mois (intervalle de 0 à 51,5+ mois ; + signale une valeur censurée). Les troubles péricardiques ont conduit à l’arrêt d’atezolizumab chez 3 (< 0,1 %) patients. Des troubles péricardiques nécessitant l’utilisation de corticoïdes sont survenues chez 0,2 % (7/5 039) des patients.
Effets de classe des inhibiteurs de point de contrôle immunitaire
Au cours du traitement par d’autres inhibiteurs de point de contrôle immunitaire, des cas de l’effet indésirable suivant, susceptibles de survenir également pendant le traitement par l’atezolizumab, ont été rapportés : insuffisance pancréatique exocrine.
Immunogénicité
Dans plusieurs essais cliniques de phase II et III, 13,1 % à 54,1 % des patients ont développé des anticorps anti-médicament (ADA) apparus sous traitement. Les patients qui ont développé des ADA en cours de traitement avaient tendance à présenter, à l’initiation, un moins bon état de santé et des caractéristiques pathologiques plus avancées. Ces déséquilibres dans l’état de santé et les caractéristiques pathologiques à l’initiation peuvent biaiser l'interprétation des analyses pharmacocinétiques (PK), d'efficacité et de sécurité. Des analyses exploratoires ajustant les déséquilibres à l’initiation sur l’état de santé et les caractéristiques pathologiques ont été menées pour évaluer l'effet des ADA sur l'efficacité. Ces analyses n'ont pas exclu une possible atténuation du bénéfice d'efficacité chez les patients ayant développé des ADA en comparaison aux patients n'ayant pas développé des ADA. Le délai médian de survenue des ADA variait de 3 semaines à 5 semaines.
Sur la base des données groupées des patients traités par atezolizumab en monothérapie (N = 3 460) et en association (N = 2 285), les fréquences suivantes d’événements indésirables ont été observées respectivement dans la population ADA positive comparée à la population ADA négative : 46,2 % d’événements indésirables de grade 3-4 vs. 39,4 %, 39,6 % d’événements indésirables graves vs. 33,3 %, 8,5 % d’événements indésirables conduisant à un arrêt de traitement vs. 7,8 % (en monothérapie) ; 63,9 % d’événements indésirables de grade 3-4 vs. 60,9 %, 43,9 % d’événements indésirables graves vs. 35,6 %, 22,8 % d’événements indésirables conduisant à un arrêt de traitement vs. 18,4 % (en association). Cependant, les données disponibles ne permettent pas de tirer des conclusions fermes sur des profils possibles d’événements indésirables.
Population pédiatrique
La sécurité d’atezolizumab chez l’enfant et l’adolescent n’a pas été établie. Aucun nouveau signal de sécurité n’a été observé lors d’un essai clinique avec 69 patients pédiatriques (< 18 ans) et le profil de sécurité était comparable aux adultes.
Personnes âgées
Aucune différence globale de sécurité n’a été observée entre les patients âgés de moins de 65 ans, de 65 à 74 ans et de 75 à 84 ans recevant l’atezolizumab en monothérapie. Les données chez les patients âgés de 85 ans et plus sont trop limitées pour tirer des conclusions significatives dans cette population.
Dans l’essai clinique IMpower150, un âge de 65 ans et plus a été associé à un risque augmenté de développer des effets indésirables chez les patients recevant l’atezolizumab en association au bevacizumab, carboplatine et paclitaxel. Dans les essais cliniques IMpower150, IMpower133 et IMpower110, les données chez les patients âgés de 75 ans et plus étaient trop limitées pour tirer des conclusions. Dans l'étude IPSOS réalisée en première ligne de traitement chez des patients atteints d’un CBNPC inéligibles à un traitement à base de sels de platine, il n'y avait aucune différence globale du profil de sécurité entre les sous-groupes d'âge des patients traités en première ligne de traitement par l’atezolizumab en monothérapie.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉROS D'AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1220/001
EU/1/17/1220/002
10. DATE DE MISE À JOUR DU TEXTE
26 mars 2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l'Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3519006 | TECENTRIQ 1200MG SOL DIL. POUR PERF FL 1 X 20ML | L01FF05 | - | € 4799,2 | Oui | - | - |
| 3983582 | TECENTRIQ 840MG SOL DIL. POUR PERF FL INJ 1X14ML | L01FF05 | - | € 3199,47 | Oui | - | - |