RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Alecensa 150 mg, gélules
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque gélule contient du chlorhydrate d’alectinib équivalent à 150 mg d’alectinib.
Excipient(s) à effet notoire:
Chaque gélule contient 33,7 mg de lactose (monohydraté) et 6 mg de sodium (laurylsulfate de sodium).
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Gélule.
Gélule blanche de 19,2 mm de longueur, portant la mention « ALE » imprimée sur la coiffe à l’encre noire et la mention « 150 mg » imprimée sur le corps à l’encre noire.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
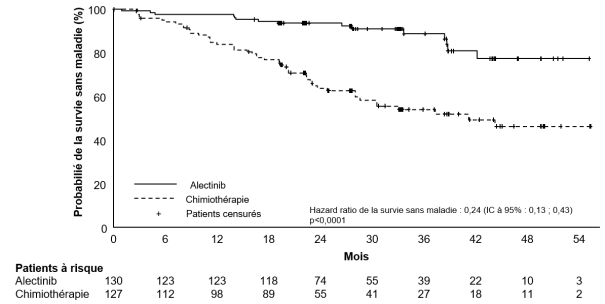
Traitement adjuvant du cancer bronchique non à petites cellules (CBNPC) réséqué
Alecensa est indiqué en monothérapie dans le traitement adjuvant, après résection complète de la tumeur, des patients adultes atteints d’un cancer bronchique non à petites cellules (CBNPC) ALK-positif à haut risque de récidive (voir rubrique 5.1 pour les critères de sélection).
Traitement du CBNPC avancé
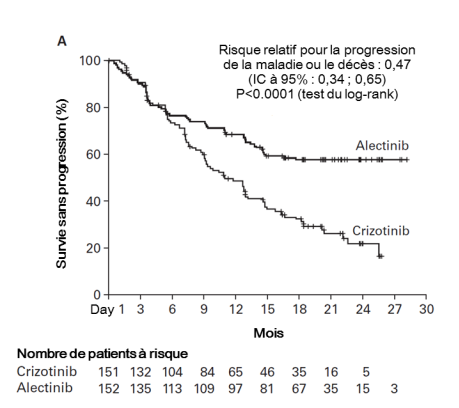
Alecensa est indiqué en monothérapie en première ligne de traitement des patients adultes atteints d’un CBNPC avancé ALK-positif.
Alecensa est indiqué en monothérapie dans le traitement du CBNPC avancé ALK-positif chez les patients adultes préalablement traités par crizotinib.
4.2 Posologie et mode d’administration
Le traitement par Alecensa doit être instauré et supervisé par un médecin expérimenté dans l’utilisation des médicaments anticancéreux.
Une méthode d’analyse d’ALK validée est nécessaire pour sélectionner les patients présentant un CBNPC avec réarrangement du gène ALK (ALK-positif). Le statut ALK-positif du CBNPC doit être établi avant l’instauration du traitement par Alecensa.
Posologie
La posologie recommandée d’Alecensa est de 600 mg (4 gélules de 150 mg) deux fois par jour au cours d’un repas (posologie quotidienne totale de 1200 mg).
Chez les patients atteints d’insuffisance hépatique sous-jacente sévère (Child-Pugh C), la posologie initiale recommandée est de 450 mg deux fois par jour au cours d’un repas (posologie quotidienne totale de 900 mg).
Durée de traitement
Traitement adjuvant du CBNPC réséqué
Le traitement par Alecensa doit être poursuivi jusqu’à récidive de la maladie, survenue d’une toxicité inacceptable ou pendant 2 ans.
Traitement du CBNPC avancé
Le traitement par Alecensa doit être poursuivi jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable.
Retard ou omission d’une dose
En cas d’omission d’une prise d’Alecensa, la dose omise doit être prise immédiatement sauf s’il reste moins de 6 heures avant la prochaine dose. Les patients ne doivent pas prendre deux doses en même temps pour compenser une dose omise. En cas de vomissement suite à une dose d’Alecensa, les patients doivent prendre la prochaine dose telle que planifiée.
Adaptations posologiques
La gestion des événements indésirables peut nécessiter une réduction de la posologie, une interruption temporaire ou un arrêt de traitement par Alecensa. La posologie d’Alecensa doit être réduite par palier de 150 mg deux fois par jour en fonction de la tolérance. Le traitement par Alecensa doit être définitivement arrêté en cas d’intolérance à la dose de 300 mg deux fois par jour.
Les recommandations d’adaptation de la posologie sont décrites dans les Tableaux 1 et 2 ci-dessous.
Tableau 1 Schéma de réduction de la posologie
Schéma de réduction de la posologie | Palier de dose |
Posologie | 600 mg deux fois par jour |
Première réduction de la posologie | 450 mg deux fois par jour |
Deuxième réduction de la posologie | 300 mg deux fois par jour |
Tableau 2 Recommandations d’adaptation de la posologie en cas d’effets indésirables spécifiques (voir rubriques 4.4 et 4.8)
Grade CTCAE | Traitement par Alecensa |
Pneumopathie interstitielle diffuse / pneumopathie de tout grade de sévérité | Arrêter immédiatement et définitivement le traitement par Alecensa si aucune autre cause de maladie pulmonaire interstitielle / pneumopathie n’a été identifiée. |
Elévation du taux d’ALAT ou d’ASAT > 5 fois la LSN avec un taux de bilirubine totale 2 fois la LSN | Interrompre temporairement le traitement jusqu’au retour à la valeur de référence ou ≤ 3 fois la LSN, puis reprendre le traitement au palier de dose inférieur (voir Tableau 1). |
Elévation du taux d’ALAT ou d’ASAT > 3 fois la LSN accompagnée d’une élévation du taux de bilirubine totale > 2 fois la LSN en l’absence de cholestase ou d’hémolyse | Arrêter définitivement le traitement par Alecensa. |
Bradycardiea de Grade 2 ou de Grade 3 (symptomatique, potentiellement sévère et médicalement significative, nécessitant une intervention médicale) | Interrompre temporairement le traitement par Alecensa jusqu’au retour à une bradycardie de Grade 1 (asymptomatique) ou à une fréquence cardiaque ≥ 60 bpm. Evaluer les médicaments concomitants connus pour entraîner une bradycardie, y compris les médicaments antihypertenseurs. |
Bradycardiea de Grade 4 (conséquences menaçant le pronostic vital, nécessitant une intervention urgente) | Arrêter définitivement le traitement par Alecensa si aucun médicament concomitant favorisant la bradycardie n’est identifié. |
Elévation des CPK > 5 fois la LSN | Interrompre temporairement le traitement par Alecensa jusqu’au retour à la valeur de référence ou à un taux ≤ 2,5 fois la LSN, puis reprendre le traitement au même palier de dose. |
Elévation des CPK > 10 fois la LSN ou deuxième élévation des CPK > 5 fois la LSN | Interrompre temporairement le traitement par Alecensa jusqu’au retour à la valeur de référence ou à un taux ≤ 2,5 fois la LSN, puis reprendre le traitement au palier de dose inférieur comme indiqué dans le Tableau 1. |
Anémie hémolytique avec un taux d’hémoglobine < 10 g/dL (Grade ≥ 2) | Interrompre temporairement le traitement par Alecensa jusqu’à résolution, puis reprendre le traitement au palier de dose inférieur (voir Tableau 1). |
ALAT = alanine aminotransférase, ASAT = aspartate aminotransférase, CPK = créatinine phosphokinase, CTCAE = National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events, LSN = limite supérieure de la normale.
a Fréquence cardiaque inférieure à 60 battements par minute (bpm).
Populations particulières
Insuffisance hépatique
Aucune adaptation de la posologie initiale n’est nécessaire chez les patients atteints d’insuffisance hépatique sous-jacente légère (Child‑Pugh A) ou modérée (Child‑Pugh B). La posologie initiale recommandée chez les patients atteints d’une insuffisance hépatique sous-jacente sévère (Child‑Pugh C) est de 450 mg deux fois par jour (posologie quotidienne totale de 900 mg) (voir rubrique 5.2). Pour tous les patients atteints d’insuffisance hépatique, une surveillance appropriée (par exemple, marqueurs de la fonction hépatique) est recommandée (voir rubrique 4.4).
Insuffisance rénale
Aucune adaptation posologique particulière n’est nécessaire chez les patients atteints d’insuffisance rénale légère à modérée. Alecensa n’a pas été étudié chez les patients atteints d’insuffisance rénale sévère. Cependant, l’élimination de l’alectinib par voie rénale étant négligeable, aucune adaptation posologique n’est nécessaire chez les patients atteints d’insuffisance rénale sévère (voir rubrique 5.2).
Patients âgés (≥ 65 ans)
Les données limitées sur la tolérance et l’efficacité d’Alecensa chez les patients âgés de 65 ans et plus ne suggèrent pas qu’une adaptation posologique soit nécessaire pour les patients âgés (voir rubrique 5.2). Aucune donnée n’est disponible pour les patients âgés de plus de 80 ans.
Population pédiatrique
La sécurité et l’efficacité d’Alecensa chez les enfants et adolescents âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Patients de très haut poids corporel (> 130 kg)
Bien que les simulations de pharmacocinétique (PK) sur Alecensa n’indiquent pas une faible exposition chez les patients de très haut poids corporel (>130 kg), alectinib est largement distribué et les études cliniques sur alectinib ont inclus des patients de poids corporel entre 36,9 et 123 kg. Aucune donnée n’est disponible pour les patients de poids corporel supérieur à 130 kg.
Mode d’administration
Alecensa est destiné à une administration orale. Les gélules doivent être avalées entières et ne doivent pas être ouvertes ou dissoutes. Elles doivent être prises au cours d’un repas (voir rubrique 5.2).
4.3 Contre-indications
Hypersensibilité à l’alectinib ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de tolérance
Les données présentées ci-dessous reflètent l’exposition à Alecensa chez 533 patients atteints d’un CBNPC réséqué ou avancé ALK-positif. Ces patients ont reçu Alecensa à la dose recommandée de 600 mg deux fois par jour dans les essais cliniques pivots pour le traitement adjuvant du CBNPC réséqué (BO40336, ALINA) ou du CBNPC avancé (BO28984, ALEX; NP28761; NP28673). Voir rubrique 5.1 pour plus d’informations sur les participants des essais cliniques.
Dans l’essai BO40336 (ALINA ; N = 128), la durée médiane d’exposition à Alecensa était de 23,9 mois. Dans l’essai BO28984 (ALEX ; N = 152), la durée médiane d’exposition à Alecensa était de 28,1 mois. Dans les essais cliniques de phase II (NP28761, NP28673 ; N = 253), la durée médiane d’exposition à Alecensa était de 11,2 mois.
Les effets indésirables les plus fréquents (≥ 20 %) étaient : constipation, myalgie, œdème, augmentation de la bilirubinémie, augmentation des ASAT, anémie, éruption cutanée et augmentation des ALAT.
Tableau des effets indésirables
Le tableau 3 liste les effets indésirables survenus chez les patients ayant reçu Alecensa au cours des essais cliniques (BO40336, BO28984, NP28761, NP28673).
Les effets indésirables listés dans le Tableau 3 sont présentés par classe de système d’organes et par catégorie de fréquence définie selon les conventions suivantes : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000). Dans chaque classe de système d’organes, les effets indésirables sont présentés par ordre décroissant de fréquence et de sévérité. Au sein du même groupe de fréquence et de sévérité, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 Effets indésirables signalés chez les patients traités par Alecensa au cours d’essais cliniques (BO40336, BO28984, NP28761, NP28673 ; N = 533).
Classe de système d’organes | Alecensa | |
| Catégorie de fréquences (tout grade) | Catégorie de fréquences (grades 3-4) |
Affections hématologiques et du système lymphatique |
|
|
Anémie1) | Très fréquent | Fréquent |
Anémie hémolytique2) | Fréquent | -* |
Affections du système nerveux |
|
|
Dysgueusie3) | Fréquent | Peu fréquent |
Affections oculaires |
|
|
Trouble de la vision4) | Fréquent | -* |
Affections cardiaques |
|
|
Bradycardie5) | Très fréquent | -* |
Affections respiratoires, thoraciques et médiastinales |
|
|
Pneumopathie interstitielle diffuse / pneumopathie inflammatoire | Fréquent | Peu fréquent |
Affections gastro-intestinales |
|
|
Diarrhée | Très fréquent | Fréquent |
Vomissements | Très fréquent | Peu fréquent |
Constipation | Très fréquent | Peu fréquent |
Nausées | Très fréquent | Peu fréquent |
Stomatite6) | Fréquent | Peu fréquent |
Affections hépatobiliaires |
|
|
Augmentation des ASAT | Très fréquent | Fréquent |
Augmentation des ALAT | Très fréquent | Fréquent |
Augmentation de la bilirubinémie7) | Très fréquent | Fréquent |
Augmentation de la phosphatase alcaline | Très fréquent | Peu fréquent |
Lésion hépatique d’origine médicamenteuse8) | Peu fréquent | Peu fréquent |
Affections de la peau et du tissu sous-cutané |
|
|
Eruption cutanée9) | Très fréquent | Fréquent |
Photosensibilité | Fréquent | Peu fréquent |
Affections musculo-squelettiques et systémiques |
|
|
Myalgie10) | Très fréquent | Peu fréquent |
Augmentation du taux sanguin de créatine phosphokinase | Très fréquent | Fréquent |
Affections du rein et des voies urinaires |
|
|
Augmentation de la créatininémie | Très fréquent | Peu fréquent** |
Lésion rénale aiguë | Fréquent | Peu fréquent** |
Troubles généraux et anomalies au site d'administration |
|
|
Œdème11) | Très fréquent | Peu fréquent |
Investigations |
|
|
Augmentation du poids | Très fréquent | Peu fréquent |
Troubles du métabolisme et de la nutrition |
|
|
Hyperuricémie12) | Fréquent | -* |
*Aucun effet indésirable de grade 3-4 n’a été observé.
**Comprend un événement de grade 5 (observé dans le CBNPC avancé).
1) comprend des cas d’anémie, de diminution de l’hémoglobine et d’anémie normochrome normocytaire.
2) des cas rapportés dans l’étude BO40336 (N = 128).
3) comprend des cas de dysgueusie, d’hypogueusie et de trouble du goût.
4) comprend des cas de vision trouble, d’atteinte visuelle, de corps flottants vitréens, de diminution de l’acuité visuelle, d’asthénopie, de diplopie, de photophobie et de photopsie.
5) comprend des cas de bradycardie et de bradycardie sinusale.
6) comprend des cas de stomatite et d’ulcération buccale.
7) comprend des cas d’augmentation du taux sanguin de bilirubine, d’hyperbilirubinémie, d’augmentation du taux sanguin de bilirubine conjuguée et d’augmentation du taux sanguin de bilirubine non conjuguée.
8) comprend deux patients avec une lésion hépatique d’origine médicamenteuse selon le terme MedDRA ainsi qu’un patient avec une augmentation des ALAT et ASAT de Grade 4 qui a une lésion hépatique d’origine médicamenteuse documentée par une biopsie hépatique.
9) comprend des cas d’éruption cutanée, d’éruption cutanée maculopapuleuse, de dermatite, de dermatite acnéiforme, d’érythème, d’éruption cutanée papuleuse, d’éruption cutanée prurigineuse, d’éruption cutanée maculeuse, d’éruption cutanée exfoliative et d’éruption érythémateuse.
10) comprend des cas de myalgie, de douleur musculo-squelettique et d’arthralgie.
11) comprend des cas d’œdème périphérique, d’œdème, d’œdème généralisé, d’œdème palpébral, d’œdème périorbital, d’œdème facial, d’œdème localisé, de gonflement périphérique, de gonflement du visage, de gonflement de la lèvre, de gonflement, de gonflement articulaire et de gonflement de la paupière.
12) comprend des cas d’hyperuricémie et d’augmentation de l’acide urique sanguin.
Description d’effets indésirables spécifiques
Pneumopathie interstitielle diffuse
Dans les essais cliniques, des cas de pneumopathie interstitielle diffuse sont survenus chez 1,7 % des patients traités par Alecensa, 0,4 % de ces cas étaient de grade 3 et des arrêts de traitement dûs à une pneumopathie interstielle diffuse sont survenus chez 1,1 % des patients, et chez 0,4 % des patients, l’événement a entraîné des modifications de la dose. Dans l’essai clinique de phase III BO28984, aucun cas de pneumopathie interstitielle diffuse de Grade 3 ou 4 n’a été observé chez les patients traités par Alecensa versus 2,0 % des patients traités par le crizotinib. Il n’y a eu aucun cas de pneumopathie interstitielle diffuse d’issue fatale dans aucun des essais cliniques. Les patients doivent faire l’objet d’une surveillance des symptômes pulmonaires évocateurs d’une pneumopathie (voir rubriques 4.2 et 4.4).
Hépatotoxicité
Dans les essais cliniques, trois patients ont présenté une lésion hépatique d’origine médicamenteuse documentée (dont deux patients pour lesquels l’effet indésirable rapporté était une lésion hépatique d’origine médicamenteuse et un patient présentant une augmentation des ASAT et ALAT de grade 4 dont l’analyse de la biopsie hépatique a conclu à une lésion hépatique d’origine médicamenteuse). Des augmentations des taux d’ASAT et d’ALAT ont été rapportées respectivement chez 23,6 % et 20,5 % des patients traités par Alecensa dans les essais cliniques. La majorité de ces événements était de Grade 1 et 2, et des événements de Grade ≥ 3 ont été rapportés chez 3,0 % des patients pour une augmentation des taux d’ASAT et 3,2 % des patients pour une augmentation des taux d’ALAT. Les événements sont généralement survenus au cours des trois premiers mois de traitement, ils étaient le plus souvent transitoires et réversibles à l’interruption temporaire du traitement par Alecensa (rapporté chez 2,3 % et 3,6 % des patients, respectivement) ou à une réduction de la posologie (1,7 % et 1,5 %, respectivement). Des augmentations des taux d’ASAT et d’ALAT, chez 1,3 % et 1,5 % des patients respectivement, ont conduit à l’arrêt du traitement par Alecensa. Des augmentations des taux d’ALAT ou d’ASAT de Grade 3 ou 4 ont été observées chez 4,6 % et 5,3 % des patients traités par Alecensa versus 16,6 % et 10,6 % des patients traités par crizotinib dans l’essai clinique de phase III BO28984.
Une augmentation du taux de bilirubine a été rapportée chez 25,9 % des patients traités par Alecensa dans les essais cliniques. La majorité des événements était d’intensité de Grade 1 et 2 ; des événements de Grade ≥ 3 ont été rapportés chez 3,9 % des patients. Ces événements sont généralement survenus au cours des trois premiers mois de traitement, ils étaient le plus souvent transitoires et la plupart étaient réversibles après modification de la posologie. Chez 8,3 % des patients, l’augmentation du taux de bilirubine a conduit à des modifications de posologie et chez 2,1 % des patients, l’augmentation du taux de bilirubine a conduit à l’arrêt du traitement par Alecensa. Dans l’essai clinique de phase III BO28984, une augmentation du taux de bilirubine de Grade 3 ou 4 est survenue chez 5,9 % des patients traités par Alecensa versus aucun patient traité par le crizotinib.
Une augmentation concomitante des ALAT ou ASAT supérieures ou égales à trois fois la LSN et de la bilirubine totale supérieure ou égale à deux fois la LSN, avec des phosphatases alcalines normales, a été rapportée chez un patient (0,2 %) traité dans les essais cliniques avec Alecensa.
Les patients doivent faire l’objet d’une surveillance de la fonction hépatique y compris les ALAT, ASAT et la bilirubine totale tel que décrit en rubrique 4.4 et doivent être pris en charge tel que recommandé en rubrique 4.2.
Bradycardie
Des cas de bradycardie (11,3 %) de Grade 1 ou 2 ont été rapportés chez des patients traités par Alecensa au cours d’essais cliniques. Aucun patient n’a présenté d’événements de Grade 3. Cent-deux des 521 patients (19,6 %) traités par Alecensa, pour lesquels des ECG en série étaient disponibles, ont eu des valeurs de fréquence cardiaque post-dose inférieures à 50 battements par minute (bpm). Dans l’essai clinique de phase III BO28984, 12,4 % des patients traités par Alecensa ont eu des valeurs de fréquence cardiaque inférieures à 50 bpm versus 17,6 % des patients traités par le crizotinib. Les patients développant une bradycardie symptomatique doivent être pris en charge tel que recommandé en rubriques 4.2 et 4.4. Aucun cas de bradycardie n’a entrainé l’arrêt du traitement par Alecensa.
Myalgie sévère et augmentation des CPK
Des cas de myalgie (35,3 %) comprenant des événements de myalgie (24,2 %), d’arthralgie (16,3 %) et des douleurs musculo-squelettiques (0,8 %) ont été rapportés chez des patients traités par Alecensa au cours des essais cliniques. La majorité des événements était de Grade 1 ou 2 et cinq patients (0,9 %) ont présenté un événement de Grade 3. Compte-tenu de ces effets indésirables, des modifications de la posologie du traitement par Alecensa ont été nécessaires pour neuf patients (1,7 %). Alecensa n’a pas été arrêté en raison de ces événements de myalgie. Une augmentation des CPK a été rapportée chez 56,2 % des 491 patients des essais cliniques pour lesquels des données biologiques sur les CPK étaient disponibles avec Alecensa. L’incidence d’une augmentation des CPK de Grade ≥ 3 était de 5,5 %. Le délai médian de survenue de l’augmentation des CPK de Grade ≥ 3 était de 15 jours dans les essais. Des modifications de dose suite à une augmentation des CPK ont été faites chez 5,4 % des patients ; aucun arrêt de traitement par Alecensa n’est survenu suite à des élévations des CPK. Dans l’essai clinique BO28984, une arthralgie sévère a été rapportée chez un patient (0,7 %) dans le bras alectinib et chez deux patients (1,3 %) dans le bras crizotinib. Une augmentation des CPK de Grade ≥ 3 a été rapportée chez 3,3 % des patients traités par Alecensa et 4,6 % des patients traités par crizotinib.
Anémie hémolytique
Une anémie hémolytique a été observée chez 3,1 % des patients traités par Alecensa au cours des essais cliniques. Ces cas étaient de grade 1 ou 2 (non graves) et n’ont pas conduit à l’arrêt du traitement (voir rubriques 4.2 et 4.4).
Effets gastro-intestinaux
Des constipations (39,6 %), des diarrhées (18,8 %), des nausées (17,6 %), et des vomissements (12,4 %) étaient les effets gastro-intestinaux (GI) les plus fréquemment rapportés. La plupart de ces événements était de sévérité faible ou modérée ; des événements de Grade 3 ont été rapportés pour les diarrhées (1,1 %), les nausées (0,4 %), les constipations (0,4 %) et les vomissements (0,2 %). Ces événements n’ont pas conduit à l’arrêt du traitement par Alecensa. Le délai médian de survenue des événements de constipations, nausées, diarrhées et/ou vomissements dans les essais cliniques était de 21 jours. La fréquence de ces effets a diminué après le premier mois de traitement. Dans l’essai clinique de phase III BO28984, des événements de Grade 3 et 4 pour les nausées et constipations ont chacun été rapportés chez un patient (0,7 %), tandis que des diarrhées ont été rapportées chez 2 patients (1,3 %) dans le bras alectinib ; l’incidence des événements de Grade 3 et 4 pour les nausées, vomissements et diarrhées était respectivement de 3,3 %, 3,3 % et 2,0 % dans le bras crizotinib.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/16/1169/001
EU/1/16/1169/002
10. DATE DE MISE À JOUR DU TEXTE
29 janvier 2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3518990 | ALECENSA 150MG CAPS DUR 4 X 56 | L01ED03 | - | € 4872,14 | Oui | - | - |