RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Kadcyla 100 mg poudre pour solution à diluer pour perfusion
Kadcyla 160 mg poudre pour solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Kadcyla 100 mg poudre pour solution à diluer pour perfusion
Un flacon de poudre pour solution à diluer pour perfusion contient 100 mg de trastuzumab emtansine. Après reconstitution, un flacon de 5 mL de solution contient 20 mg/mL de trastuzumab emtansine (voir rubrique 6.6).
Kadcyla 160 mg poudre pour solution à diluer pour perfusion
Un flacon de poudre pour solution à diluer pour perfusion contient 160 mg de trastuzumab emtansine. Après reconstitution, un flacon de 8 mL de solution contient 20 mg/mL de trastuzumab emtansine (voir rubrique 6.6).
Excipients à effet notoire
Chaque flacon de 100 mg contient 1,38 mg de sodium et 1,1 mg de polysorbate 20.
Chaque flacon de 160 mg contient 2,24 mg de sodium et 1,7 mg de polysorbate 20.
Pour la liste complète des excipients, voir rubrique 6.1.
Le trastuzumab emtansine est un anticorps conjugué qui contient du trastuzumab, un anticorps monoclonal humanisé recombinant de classe IgG1 produit par une culture de cellules de mammifère (ovaire de hamster chinois), lié de façon covalente au DM1, un inhibiteur de microtubules, grâce à l’agent de liaison thioéther stable MCC (4-[N-maleimidométhyl] cyclohexane-1-carboxylate).
3. FORME PHARMACEUTIQUE
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée blanche à blanc cassé.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Cancer du sein précoce
Kadcyla, en monothérapie, est indiqué dans le traitement adjuvant de patients adultes atteints d’un cancer du sein précoce HER2 positif qui présentent une maladie résiduelle invasive, au niveau du sein et/ou des ganglions lymphatiques, après un traitement néoadjuvant à base de taxane et d’un traitement anti-HER2.
Cancer du sein métastatique
Kadcyla, en monothérapie, est indiqué dans le traitement de patients adultes atteints d’un cancer du sein HER2 positif métastatique ou localement avancé non résécable, ayant reçu au préalable du trastuzumab et un taxane, séparément ou en association. Les patients doivent :
- avoir reçu un traitement antérieur pour la maladie localement avancée ou métastatique, ou
- avoir présenté une progression de la maladie pendant un traitement adjuvant ou dans les six mois suivant sa fin.
4.2 Posologie et mode d’administration
Kadcyla doit être prescrit uniquement par un médecin et administré en perfusion intraveineuse sous la
surveillance d’un professionnel de santé expérimenté dans le traitement de patients atteints de cancer
(c’est-à-dire prêt à prendre en charge des réactions allergiques/anaphylactiques liées à la perfusion et
dans un environnement où un équipement complet de réanimation est immédiatement disponible (voir
rubrique 4.4)).
Les patients traités avec le trastuzumab emtansine doivent présenter un statut tumoral HER2 positif, défini par un score 3+ par immunohistochimie (IHC) ou un ratio ≥ 2,0 par hybridation in situ (HIS) ou par hybridation in situ en fluorescence (FISH), déterminé par un dispositif médical de Diagnostic In Vitro (DIV) marqué CE. Si un DIV marqué CE n’est pas disponible, le statut HER2 doit être déterminé par une méthode alternative validée.
Afin d’éviter des erreurs médicamenteuses, il est important de vérifier les étiquettes du flacon afin de s’assurer que le médicament préparé et administré est Kadcyla (trastuzumab emtansine) et non un autre médicament contenant du trastuzumab (par ex. trastuzumab ou trastuzumab déruxtécan).
Posologie
La dose recommandée de trastuzumab emtansine est de 3,6 mg/kg de poids corporel administrée en perfusion intraveineuse toutes les trois semaines (cycle de 21 jours).
La dose initiale doit être administrée en perfusion intraveineuse de 90 minutes. Les patients doivent être surveillés pendant la perfusion et pendant au moins 90 minutes après la fin de la perfusion initiale pour des symptômes de fièvre, frissons ou d’autres réactions liées à la perfusion. Le site de perfusion doit être étroitement surveillé pour détecter une possible infiltration sous-cutanée pendant l’administration. Des cas d’atteinte épidermique ou de nécrose retardées suite à une extravasation ont été observés depuis la commercialisation (voir rubriques 4.4 et 4.8).
Si la perfusion précédente a été bien tolérée, les doses suivantes de trastuzumab emtansine peuvent être administrées en perfusions de 30 minutes. Les patients doivent être surveillés pendant la perfusion et pendant au moins 30 minutes après la fin de la perfusion.
La vitesse de perfusion du trastuzumab emtansine doit être diminuée ou la perfusion doit être interrompue si le patient développe des symptômes liés à la perfusion (voir rubriques 4.4 et 4.8). Le traitement par trastuzumab emtansine doit être arrêté en cas de réactions liées à la perfusion menaçant le pronostic vital.
Durée du traitement
Cancer du sein précoce
Les patients doivent recevoir le traitement pendant une période totale de 14 cycles, sauf en cas de rechute de la maladie ou de survenue d’une toxicité non contrôlable.
Cancer du sein métastatique
Les patients doivent recevoir le traitement jusqu’à la progression de la maladie ou survenue d’une toxicité non contrôlable.
Modification de dose
Le traitement des effets indésirables symptomatiques peut nécessiter une interruption temporaire du traitement, une réduction de la dose ou l’arrêt du traitement par trastuzumab emtansine conformément aux recommandations présentées ci-dessous et dans les tableaux 1 et 2.
La dose de trastuzumab emtansine ne doit pas être ré-augmentée après qu’une réduction de dose ait été effectuée.
Tableau 1 Schéma de réduction de dose
Schéma de réduction de dose | Dose à administrer |
Première réduction de dose | 3 mg/kg |
Deuxième réduction de dose | 2,4 mg/kg |
Nécessité d’une réduction de dose supplémentaire | Arrêt du traitement |
Tableau 2 Recommandations de modification de dose
Modifications de dose dans le cancer du sein précoce | ||
Effet indésirable | Sévérité | Modification du traitement |
Thrombocytopénie | Grade 2-3 le jour de traitement prévu | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade 1 (≥ 75 000/mm3), puis reprendre le traitement au même niveau de dose. Si un patient nécessite 2 reports de traitement en raison d’une thrombocytopénie, envisager de réduire la dose d’un niveau. |
Grade 4 à tout moment | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade 1 (≥ 75 000/mm3), puis reprendre le traitement en réduisant la dose d’un niveau. | |
Augmentation du taux d’alanine aminotransférase (ALAT) | Grade 2-3 (> 3,0 à ≤ 20 x la LSN le jour de traitement prévu) | Ne pas administrer le trastuzumab emtansine avant que le taux d’ALAT ne revienne à un grade ≤ 1, puis reprendre le traitement en réduisant la dose d’un niveau. |
Grade 4 | Arrêter le traitement avec le trastuzumab emtansine. | |
Augmentation du taux d’aspartate aminotransférase (ASAT) | Grade 2 | Ne pas administrer le trastuzumab emtansine avant que le taux d’ASAT ne revienne à un grade ≤ 1, puis reprendre le traitement au même niveau de dose. |
Grade 3 | Ne pas administrer le trastuzumab emtansine avant que le taux d’ASAT ne revienne à un grade ≤ 1, puis reprendre le traitement en réduisant la dose d’un niveau. | |
Grade 4 | Arrêter le traitement avec le trastuzumab emtansine. | |
Hyperbilirubinémie | BILIT > 1,0 à ≤ 2,0 x la LSN le jour de traitement prévu | Ne pas administrer le trastuzumab emtansine avant que le taux de bilirubine totale ne revienne à ≤ 1,0 × la LSN, puis reprendre le traitement en réduisant la dose d’un niveau. |
BILIT > 2 x la LSN à tout moment | Arrêter le traitement avec le trastuzumab emtansine. | |
Atteinte hépatique médicamenteuse (DILI) | Transaminases sériques > 3 x la LSN et bilirubine totale concomitante > 2 xla LSN | Arrêter définitivement le traitement avec le trastuzumab emtansine en l’absence d’autre cause probable de l’élévation des enzymes hépatiques et de la bilirubine, par ex. métastases hépatiques ou médicament concomitant. |
Hyperplasie nodulaire régénérative (HNR) | Tous grades | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
Neuropathie périphérique | Grade 3-4 | Ne pas administrer le trastuzumab emtansine avant amélioration jusqu’à un grade 2. |
Dysfonctionnement ventriculaire gauche | FEVG < 45 % | Ne pas administrer le trastuzumab emtansine. |
FEVG 45 % à < 50 % et diminution ≥ 10 points de la valeur initiale* | Ne pas administrer le trastuzumab emtansine. | |
FEVG 45 % à < 50 % et diminution < 10 points de la valeur initiale* | Continuer le traitement avec le trastuzumab emtansine. Répéter l’évaluation de la FEVG dans les 3 semaines. | |
FEVG ≥ 50 % | Continuer le traitement avec le trastuzumab emtansine. | |
Insuffisance cardiaque | ICC symptomatique, | Arrêter le traitement avec le trastuzumab emtansine. |
Toxicité pulmonaire | Pneumopathie interstitielle diffuse ou pneumopathie | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
Pneumopathie radique | Grade 2 | Arrêter le traitement avec le trastuzumab emtansine en l’absence de résolution avec le traitement standard. |
Grade 3-4 | Arrêter le traitement avec le trastuzumab emtansine. | |
Modifications de dose dans le cancer du sein métastatique | ||
Effet indésirable | Sévérité | Modification du traitement |
Thrombocytopénie | Grade 3 | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade 1 (≥ 75 000/mm3), puis reprendre le traitement au même niveau de dose. |
Grade 4 | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade 1 (≥ 75 000/mm3), puis reprendre le traitement en réduisant la dose d’un niveau. | |
Augmentation des transaminases (ASAT/ALAT) | Grade 2 ( 2,5 à ≤ 5 x la LSN) | Continuer le traitement au même niveau de dose. |
Grade 3 ( 5 à ≤ 20 la LSN) | Ne pas administrer le trastuzumab emtansine avant que le taux d’ASAT/ALAT ne revienne à un grade ≤ 2, puis reprendre le traitement en réduisant la dose d’un niveau. | |
Grade 4 ( 20 xla LSN) | Arrêter le traitement avec le trastuzumab emtansine. | |
Hyperbilirubinémie | Grade 2 | Ne pas administrer le trastuzumab emtansine avant que le taux de bilirubine totale ne revienne à un grade ≤ 1, puis reprendre le traitement au même niveau de dose. |
Grade 3 | Ne pas administrer le trastuzumab emtansine avant que le taux de bilirubine totale ne revienne à un grade ≤ 1, puis reprendre le traitement en réduisant la dose d’un niveau. | |
Grade 4 | Arrêter le traitement avec le trastuzumab emtansine. | |
Atteinte hépatique médicamenteuse (DILI) | Transaminases sériques > 3 x la LSN et bilirubine totale concomitante > 2 x la LSN | Arrêter définitivement le traitement avec le trastuzumab emtansine en l’absence d’autre cause probable de l’élévation des enzymes hépatiques et de la bilirubine, par ex. métastases hépatiques ou médicament concomitant. |
Hyperplasie nodulaire régénérative (HNR) | Tous grades | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
Dysfonctionnement ventriculaire gauche | ICC symptomatique | Arrêter le traitement avec le trastuzumab emtansine. |
FEVG < 40 % | Ne pas administrer le trastuzumab emtansine. | |
FEVG 40 % à ≤ 45 % et diminution ≥ 10 points de la valeur initiale | Ne pas administrer le trastuzumab emtansine. | |
FEVG 40 % à ≤ 45 % et diminution < 10 points de la valeur initiale | Continuer le traitement avec le trastuzumab emtansine. Répéter l’évaluation de la FEVG dans les 3 semaines. | |
FEVG > 45 % | Continuer le traitement avec le trastuzumab emtansine. | |
Neuropathie périphérique | Grade 3-4 | Ne pas administrer le trastuzumab emtansine avant amélioration jusqu’à un grade 2. |
Toxicité pulmonaire | Pneumopathie interstitielle diffuse ou pneumopathie | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
ALAT alanine aminotransférase ; ASAT aspartate aminotransférase ; ICC = insuffisance cardiaque congestive ; FEVG fraction d’éjection ventriculaire gauche ; DSVG dysfonctionnement systolique ventriculaire gauche ; BILIT = bilirubine totale ; LSN limite supérieure de la normale
* Avant le début du traitement par le trastuzumab emtansine.
Oubli ou retard de dose
Si une dose programmée n’est pas administrée, elle doit être administrée dès que possible, sans attendre le prochain cycle prévu. Le calendrier d’administration doit être modifié afin de maintenir un intervalle de 3 semaines entre les doses. La dose suivante doit être administrée conformément aux recommandations de posologie ci-dessus.
Neuropathie périphérique
Le traitement avec le trastuzumab emtansine doit être interrompu de façon temporaire chez les patients présentant une neuropathie périphérique de grade 3 ou 4 jusqu’à amélioration à un grade ≤ 2. Lors de la reprise du traitement, une réduction de dose peut être envisagée selon le schéma de réduction de dose (voir tableau 1).
Populations particulières
Patients âgés
Aucune adaptation de dose n’est requise chez les patients âgés de 65 ans et plus. Les données sont insuffisantes pour établir la sécurité et l’efficacité chez les patients âgés de 75 ans et plus du fait des données limitées dans ce sous-groupe. Cependant, chez les patients âgés de 65 ans et plus, une analyse d’un sous-groupe de 345 patients de l’étude MO28231 montre une tendance à des incidences plus élevées d’effets indésirables de grade 3, 4 et 5, d’effets indésirables graves et d’effets indésirables conduisant à un arrêt ou une interruption du traitement, mais avec une incidence similaire d’effets indésirables de grade 3 et plus classés comme liés au traitement.
Une analyse pharmacocinétique de population montre que l’âge n’a pas d’effet cliniquement significatif sur la pharmacocinétique du trastuzumab emtansine (voir rubriques 5.1 et 5.2).
Insuffisance rénale
Aucune adaptation de la dose initiale n’est requise chez les patients présentant une insuffisance rénale légère ou modérée (voir rubrique 5.2). Le besoin potentiel d’une adaptation de dose chez les patients présentant une insuffisance rénale sévère ne peut être déterminé en raison de l’insuffisance des données. Par conséquent, les patients avec une insuffisance rénale sévère doivent être étroitement surveillés.
Insuffisance hépatique
Aucune adaptation de la dose initiale n’est requise chez les patients présentant une insuffisance hépatique légère ou modérée. Le trastuzumab emtansine n’a pas été étudié chez les patients avec une insuffisance hépatique sévère. Le traitement des patients insuffisants hépatiques doit être initié avec précaution en raison de l’hépatotoxicité observée avec le trastuzumab emtansine (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. Il n’existe pas d’utilisation justifiée dans la population pédiatrique dans l’indication de cancer du sein.
Mode d’administration
Kadcyla est à administrer par voie intraveineuse. Le trastuzumab emtansine doit être reconstitué et dilué par un professionnel de santé et administré en perfusion intraveineuse. Il ne doit pas être administré en injection rapide ou bolus intraveineux.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
La sécurité du trastuzumab emtansine a été évaluée chez 2 611 patients atteints d’un cancer du sein dans les études cliniques. Dans cette population de patients :
- les effets indésirables graves les plus fréquents (> 0,5 % des patients) étaient hémorragie, fièvre, thrombocytopénie, dyspnée, douleurs abdominales, douleurs musculosquelettiques et vomissements.
- les effets indésirables les plus fréquents (≥ 25 %) avec le trastuzumab emtansine étaient nausées, fatigue, douleurs musculosquelettiques, hémorragie, céphalées, augmentation des transaminases, thrombocytopénie et neuropathie périphérique. La majorité des effets indésirables rapportés était d’une sévérité de grade 1 ou 2.
- les effets indésirables de grade ≥ 3 selon les critères Common Terminology Criteria for Adverse Events du National Cancer Institute (NCI-CTCAE) les plus fréquentes (> 2 %) étaient : thrombocytopénie, augmentation des transaminases, anémie, neutropénie, fatigue et hypokaliémie.
Tableau des effets indésirables
Les effets indésirables chez 2 611 patients traités avec le trastuzumab emtansine sont présentées dans le tableau 3. Les effets indésirables sont listées ci-dessous selon la classification MedDRA des classes de systèmes d’organes et les catégories de fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence et de classe de systèmes d’organes, les effets indésirables sont présentées suivant un ordre décroissant de gravité. Les effets indésirables ont été rapportées selon les critères NCI-CTCAE pour l’évaluation de la toxicité.
Tableau 3 Tableau des effets indésirables chez les patients traités par trastuzumab emtansine dans les études cliniques
Classe de systèmes d’organes | Fréquence | Effets indésirables |
Infections et infestations | Très fréquent | Infection urinaire |
Affections hématologiques et du système lymphatique | Très fréquent | Thrombocytopénie, anémie |
Fréquent | Neutropénie, leucopénie | |
Affections du système immunitaire | Fréquent | Réaction d’hypersensibilité |
Troubles du métabolisme et de la nutrition | Fréquent | Hypokaliémie |
Affections psychiatriques | Très fréquent | Insomnie |
Affections du système nerveux | Très fréquent | Neuropathie peripherique, céphalées |
Fréquent | Vertiges, dysgueusie, troubles de la mémoire | |
Affections oculaires | Fréquent | Sécheresse oculaire, conjonctivite, vision floue, augmentation du larmoiement |
Affections cardiaques | Fréquent | Dysfonctionnement ventriculaire gauche |
Affections vasculaires | Très fréquent | Hémorragie |
Fréquent | Hypertension | |
Affections respiratoires, thoraciques et médiastinales | Très fréquent | Epistaxis, toux, dyspnée |
Peu fréquent | Pneumopathie (pneumopathie interstitielle diffuse) | |
Affections gastro-intestinales | Très fréquent | Stomatite, diarrhée, vomissements, nausées, constipation, sécheresse buccale, douleurs abdominales |
Fréquent | Dyspepsie, saignement gingival | |
Affections hépatobiliares | Très fréquent | Augmentation des transaminases |
Fréquent | Augmentation des phosphatases alcalines sanguines, augmentation de la bilirubine sanguine | |
Peu fréquent | Hépatotoxicité, hyperplasie nodulaire régénérative, hypertension portale | |
Rare | Insuffisance hépatique | |
Affections de la peau et du tissu sous-cutané | Fréquent | Rash, prurit, alopécie, trouble unguéal, syndrome d’érythrodysesthésie palmo-plantaire, urticaire |
Affections musculosquelettiques et systémiques | Très fréquent | Douleurs musculosquelettiques, arthralgie, myalgie |
Troubles généraux et anomalies au site d’administration | Très fréquent | Fatigue, fièvre, asthénie |
Fréquent | Œdème périphérique, frissons | |
Peu fréquent | Extravasation au site d’injection | |
Lésions, intoxications et complications liées aux procédures | Fréquent | Réactions liées à la perfusion |
Peu fréquent | Pneumopathie radique |
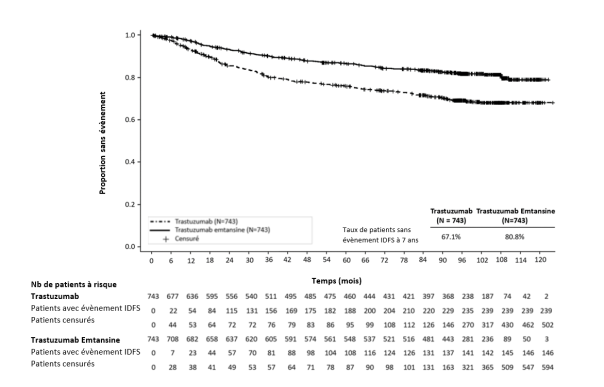
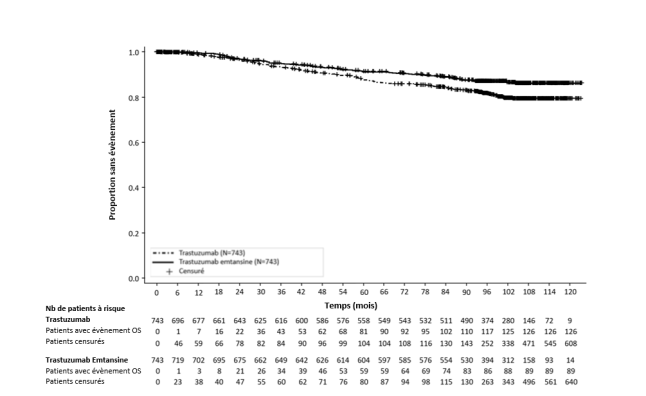
Le tableau 3 présente les données groupées de la période totale de traitement dans les études cliniques dans le cancer du sein métastatique (N = 1 871 ; le nombre médian de cycles de trastuzumab emtansine était de 10) et dans l’étude clinique KATHERINE (N = 740 ; le nombre médian de cycles était de 14).
Description des effets indésirables spécifiques
Thrombocytopénie
Une thrombocytopénie ou une diminution du taux de plaquettes a été rapportée chez 24,9 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine et a été la réaction indésirable la plus fréquente conduisant à un arrêt du traitement (2,6 %). Une thrombocytopénie a été rapportée chez 28,6 % des patients dans les études cliniques dans le cancer du sein précoce avec le trastuzumab emtansine et a été la réaction indésirable la plus rapportée tous grades confondus et pour les grades ≥ 3, ainsi que la réaction indésirable ayant mené au plus grand nombre d’arrêts de traitement (4,2 %), d’interruptions de traitement et de réductions de doses. La majorité des patients a présenté des évènements de grade 1 ou 2 (≥ 50 000/mm3), avec un nadir survenant jusqu’au jour 8 et revenant généralement à un grade 0 ou 1 (≥ 75 000/mm3) avant la dose programmée suivante. Dans les études cliniques, l’incidence et la sévérité de la thrombocytopénie étaient plus élevées chez les patients asiatiques. Indépendamment de la race, l’incidence des évènements de grade 3 ou 4 (< 50 000/mm3) était de 8,7 % chez les patients atteints d’un cancer du sein métastatique traités avec le trastuzumab emtansine et de 5,7 % chez les patients atteints d’un cancer du sein précoce. Pour les modifications de dose en cas de thrombocytopénie, voir rubriques 4.2 et 4.4.
Hémorragie
Des évènements hémorragiques ont été rapportés chez 34,8 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine et l’incidence des évènements hémorragiques sévères (grade ≥ 3) a été de 2,2 %. Des évènements hémorragiques ont été rapportés chez 29,2 % des patients atteints d’un cancer du sein précoce et l’incidence des évènements hémorragiques sévères (grade ≥ 3) a été de 0,4 %, dont un évènement de grade 5. Parmi les cas observés, certains patients avaient une thrombocytopénie ou recevaient également un traitement anticoagulant ou antiagrégant plaquettaire, d’autres n’avaient aucun facteur de risque supplémentaire connu. Des évènements hémorragiques avec une issue fatale ont été observés pour les patients atteints d’un cancer du sein métastatique comme pour ceux atteints d’un cancer du sein précoce.
Augmentation des transaminases (ASAT/ALAT)
Une augmentation des transaminases sériques (grade 1 - 4) a été observée pendant le traitement par trastuzumab emtansine dans les études cliniques (voir rubrique 4.4). L’élévation des transaminases était généralement transitoire. Un effet cumulatif du trastuzumab emtansine sur les transaminases a été observé, avec généralement un retour à la normale après l’arrêt du traitement. Des augmentations des transaminases ont été rapportées chez 24,2 % des patients dans les études cliniques dans le cancer du sein métastatique. Des augmentations des taux d’ASAT et d’ALAT de grade 3 ou 4 ont été rapportées chez respectivement 4,2 % et 2,7 % des patients atteints d’un cancer du sein métastatique et sont généralement survenues dans les premiers cycles du traitement (1 - 6). Des augmentations des transaminases ont été rapportées chez 32,6 % des patients atteints d’un cancer du sein précoce. Des augmentations des transaminases de grade 3 et 4 ont été rapportées chez 1,6 % des patients atteints d’un cancer du sein précoce. En général, la survenue d’événements hépatiques de grade ≥ 3 n’était pas associée à une détérioration de l’état clinique. Les valeurs de suivi ultérieures tendaient à montrer une amélioration à des intervalles autorisant le patient à continuer l’étude et à continuer à recevoir le traitement de l’étude à la même dose ou à une dose réduite. Aucune relation n’a été observée entre l’exposition au trastuzumab emtansine (ASC), la concentration sérique maximale de trastuzumab emtansine (Cmax), l’exposition totale au trastuzumab (ASC) ou la Cmax du DM1 et l’augmentation des transaminases. Pour les modifications de dose en cas d’augmentation des transaminases, voir rubriques 4.2 et 4.4.
Dysfonctionnement ventriculaire gauche
Un dysfonctionnement ventriculaire gauche a été rapporté chez 2,2 % des patients atteints d’un cancer du sein métastatique dans les études cliniques avec le trastuzumab emtansine. La majorité des évènements était une diminution de la FEVG de grade 1 ou 2 asymptomatique. Des évènements de grade 3 ou 4 ont été rapportés chez 0,4 % des patients atteints d’un cancer du sein métastatique. Dans une étude observationnelle (BO39807), environ 22 % (7 sur 32) des patients atteints d’un cancer du sein métastatique débutant un traitement par trastuzumab emtansine avec une FEVG de 40 – 49 % à l’état initial, ont présenté une diminution de la FEVG > 10 points de la valeur initiale et/ou une insuffisance cardiaque congestive (ICC) ; la plupart de ces patients présentaient d’autres facteurs de risque cardiovasculaire. Un dysfonctionnement ventriculaire gauche est survenu chez 3,0 % des patients atteints d’un cancer du sein précoce, avec un grade 3 chez 0,5 % des patients et aucun évenements de grade plus élevé. Pour les modifications de dose en cas de diminution de la FEVG, voir le tableau 2 à la rubrique 4.2 et la rubrique 4.4.
Neuropathie périphérique
Une neuropathie périphérique, principalement de grade 1 et à prédominance sensorielle, a été rapportée dans les études cliniques avec le trastuzumab emtansine. Chez les patients atteints d’un cancer du sein métastatique, l’incidence globale des neuropathies périphériques était de 29,0 % et de 8,6 % pour les grades ≥ 2. Chez les patients atteints d’un cancer du sein précoce, l’incidence globale était de 32,0 % et de 10,1 % pour les grades ≥ 2.
Réactions liées à la perfusion
Les réactions liées à la perfusion sont caractérisées par un ou plusieurs des symptômes suivants : bouffées de chaleur, frissons, fièvre, dyspnée, hypotension, râles sibilants, bronchospasme et tachycardie. Des réactions liées à la perfusion ont été rapportées chez 4,0 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine, avec six évènements de grade 3 et aucun évènement de grade 4 rapportés. Des réactions liées à la perfusion ont été rapportées chez 1,6 % des patients atteints d’un cancer du sein précoce, sans aucun évènement de grade 3 ou 4 rapporté. Les réactions liées à la perfusion se sont résolues en quelques heures à un jour après la fin de la perfusion. Aucun effet de dose n’a été observé dans les études cliniques. Pour les modifications de dose en cas de réactions liées à la perfusion, voir rubriques 4.2 et 4.4.
Réactions d’hypersensibilité
Une hypersensibilité a été rapportée chez 2,6 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine, avec un évènement de grade 3 et un évènement de grade 4 rapportés. Une hypersensibilité a été rapportée chez 2,7 % des patients atteints d’un cancer du sein précoce, avec un grade 3 chez 0,4 % des patients et aucun évenements de grade plus élevé. Généralement, la majorité des réactions d’hypersensibilité était d’une sévérité légère ou modérée et s’est résolue après traitement. Pour les modifications de dose en cas de réactions d’hypersensibilité, voir rubriques 4.2 et 4.4.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe un risque de réponse immunitaire au trastuzumab emtansine. Un total de 1 243 patients issus de sept études cliniques a été testé à plusieurs intervalles de temps pour des anticorps anti-médicament au trastuzumab emtansine. Après administration du trastuzumab emtansine, 5,1 % (64/1 243) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine à un ou plusieurs intervalles de temps post-administration. Dans les études de phases I et II, 6,4 % (24/376) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine. Dans l’étude EMILIA (TDM4370g/BO21977), 5,2 % (24/466) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine, dont 13 étaient également positifs pour des anticorps neutralisants. Dans l’étude KATHERINE (BO27938), 4,0 % (16/401) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine, dont 5 étaient également positifs pour des anticorps neutralisants. En raison de la faible incidence des anticorps anti-médicament, l’impact des ces anticorps sur la pharmacocinétique, la pharmacodynamie, la sécurité et/ou l’efficacité du trastuzumab emtansine n’est pas connu.
Extravasation
Des réactions secondaires à l’extravasation ont été observées dans les études cliniques avec le trastuzumab emtansine. Ces réactions étaient généralement légères ou modérées et comprenaient un érythème, une sensibilité, une irritation de la peau, une douleur ou un gonflement au site de perfusion. Ces réactions ont été observées plus fréquemment dans les 24 heures suivant la perfusion. Depuis la commercialisation, des cas d’atteinte épidermique ou de nécrose suite à une extravasation ont été exceptionnellement observés dans les jours ou les semaines après la perfusion. Aucun traitement spécifique de l’extravasation du trastuzumab emtansine n’est connu à ce jour (voir rubrique 4.4).
Anomalies biologiques
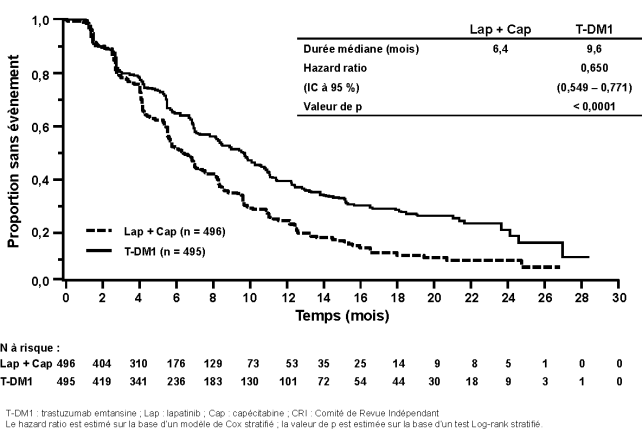
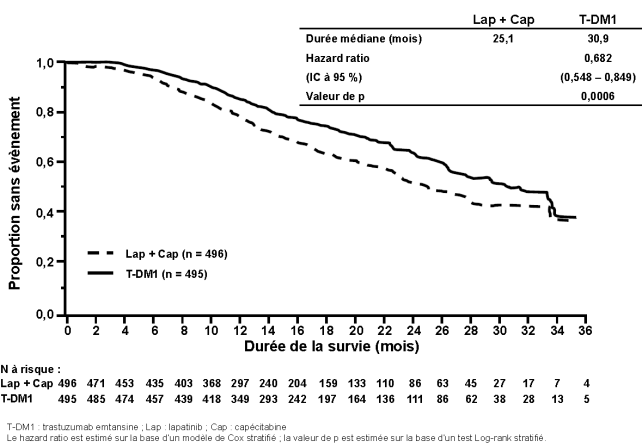
Les tableaux 4 et 5 présentent les anomalies biologiques observées chez les patients traités avec le trastuzumab emtansine dans l’étude clinique TDM4370g/BO21977/EMILIA et l’étude clinique BO27938/KATHERINE.
Tableau 4 Anomalies biologiques observées chez les patients traités avec le trastuzumab
emtansine dans l’étude clinique TDM4370g/BO21977/EMILIA
Paramètre | Trastuzumab emtansine (N = 490) | ||
Tous grades (%) | Grade 3 (%) | Grade 4 (%) | |
Hépatique | |||
Taux de bilirubine augmenté | 21 | < 1 | 0 |
Taux d’ASAT augmenté | 98 | 8 | < 1 |
Taux d’ALAT augmenté | 82 | 5 | < 1 |
Hématologique | |||
Taux de plaquettes diminué | 85 | 14 | 3 |
Taux d’hémoglobine diminué | 63 | 5 | 1 |
Taux de neutrophiles diminué | 41 | 4 | < 1 |
Potassium | |||
Taux de potassium diminué | 35 | 3 | < 1 |
Tableau 5 Anomalies biologiques observées chez les patients traités avec le trastuzumab emtansine dans l’étude clinique BO27938/KATHERINE
Paramètre | Trastuzumab emtansine (N = 740) | ||
Tous grades (%) | Grade 3 (%) | Grade 4 (%) | |
Hépatique | |||
Taux de bilirubine augmenté | 11 | 0 | 0 |
Taux d’ASAT augmenté | 79 | < 1 | 0 |
Taux d’ALAT augmenté | 55 | < 1 | 0 |
Hématologique | |||
Taux de plaquettes diminué | 51 | 4 | 2 |
Taux d’hémoglobine diminué | 31 | 1 | 0 |
Taux de neutrophiles diminué | 24 | 1 | 0 |
Potassium | |||
Taux de potassium diminué | 26 | 2 | < 1 |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/13/885/001
EU/1/13/885/002
10. DATE DE MISE À JOUR DU TEXTE
20 février 2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3084670 | KADCYLA 160 MG PULV SOL DIL PERF 20 MG/ML | L01FD03 | - | € 2217,26 | Oui | - | - |
| 3084688 | KADCYLA 100 MG PULV SOL DIL PERF 20 MG/ML | L01FD03 | - | € 1385,79 | Oui | - | - |