RESUME DES CARACTERISTIQUES DU PRODUIT
1. DENOMINATION DU MEDICAMENT
Zelboraf 240 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé contient 240 mg de vemurafenib (sous forme de coprécipité de vemurafenib et d’acétyl succinate d'hypromellose).
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé).
Comprimé pelliculé blanc rosâtre à blanc orangé, ovale, biconvexe, d’environ 19 mm avec inscription ‘VEM’ sur une face.
4. DONNEES CLINIQUES
4.1 Indication thérapeutique
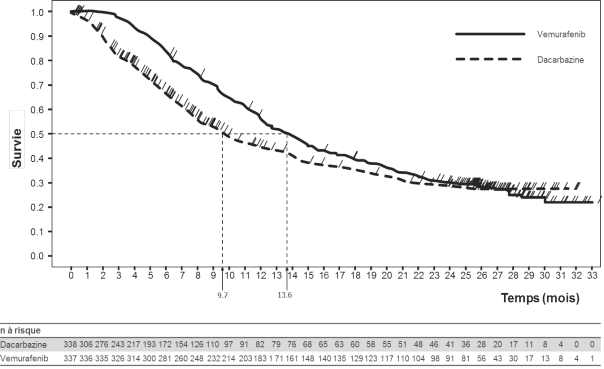
Le vemurafenib est indiqué en monothérapie dans le traitement des patients adultes atteints d’un mélanome non résécable ou métastatique porteur d’une mutation BRAF V600 (voir rubrique 5.1).
4.2 Posologie et mode d’administration
Le traitement par le vemurafenib doit être initié et supervisé par un médecin qualifié expérimenté dans l'utilisation des traitements anticancéreux.
Avant le début du traitement par le vemurafenib, la présence de la mutation BRAF V600 doit être confirmée par un test validé (voir rubriques 4.4 et 5.1).
Posologie
La dose recommandée de vemurafenib est de 960 mg (soit 4 comprimés à 240 mg) deux fois par jour (soit une dose quotidienne totale de 1920 mg). Le vemurafenib peut être pris avec ou sans nourriture, toutefois la prise à jeun des deux doses quotidiennes de manière constante doit être évitée (voir la section 5.2).
Durée du traitement
Le traitement par le vemurafenib doit être poursuivi jusqu’à progression de la maladie ou survenue d’une toxicité inacceptable (voir tableaux 1 et 2 ci-dessous).
Omission d’une dose
Si une dose est omise, elle peut être prise jusqu’à 4 heures avant la dose suivante afin de maintenir la fréquence d’administration à deux prises par jour. Les deux doses ne doivent pas être prises simultanément.
Vomissement
En cas de vomissement suite à l’administration du vemurafenib, le patient ne doit pas prendre de dose supplémentaire et le traitement doit être poursuivi de manière habituelle.
Adaptations posologiques
La prise en charge des effets indésirables ou d’un allongement de l’intervalle QTc peut nécessiter une réduction de dose, une interruption temporaire et/ou un arrêt du traitement (voir tableaux 1et 2). Il est déconseillé d’effectuer des adaptations posologiques conduisant à la prise de moins de 480 mg deux fois par jour.
En cas de survenue d’un carcinome épidermoïde cutané (CEC), il est recommandé de poursuivre le traitement sans modification de la dose du vemurafenib (voir rubriques 4.4 et 4.8).
Tableau 1 : Schéma d’adaptation posologique selon le grade des effets indésirables
Grade (CTC-AE)(a) | Modification de posologie recommandée |
Grade 1 ou grade 2 (tolérable) | Maintien de la dose de vemurafenib à 960 mg deux fois par jour. |
Grade 2 (intolérable) ou grade 3 |
|
1ère apparition d’un effet indésirable de grade 2 ou 3 | Interruption du traitement jusqu’au retour à un grade 0 ou 1. Reprise du traitement à 720 mg deux fois par jour (ou à 480 mg deux fois par jour si la dose a déjà été réduite). |
2ème apparition d’un effet indésirable de grade 2 ou 3 ou persistance après interruption du traitement | Interruption du traitement jusqu’au retour à un grade 0 ou 1. Reprise du traitement à 480 mg deux fois par jour (ou arrêt définitif si la dose a déjà été réduite à 480 mg deux fois par jour). |
3ème apparition d’un effet indésirable de grade 2 ou 3 ou persistance après une seconde réduction de dose | Arrêt définitif. |
Grade 4 |
|
1ère apparition d’un effet indésirable de grade 4 | Arrêt définitif ou interruption du traitement par le vemurafenib jusqu’au retour à un grade 0 ou 1. |
2nde apparition d’un effet indésirable de grade 4 ou persistance d’un effet indésirable de grade 4 après une première réduction de dose | Arrêt définitif. |
(a) Intensité des événements indésirables cliniques cotée selon les critères communs de terminologie pour les événements indésirables (Common Terminology Criteria for Adverse Events ; CTC-AE) v4.0.
Un allongement de l’intervalle QT exposition-dépendante a été observé lors d’une étude de phase II, non contrôlée, menée en ouvert chez des patients atteints d’un mélanome métastatique ayant été préalablement traités. La prise en charge d’un allongement de l’intervalle QT peut nécessiter des mesures de surveillance spécifiques (voir rubrique 4.4).
Tableau 2: Schéma d’adaptation posologique en fonction de l’allongement de l’intervalle QT
Valeur du QTc | Modification de posologie recommandée |
QTc>500 ms avant traitement | Traitement déconseillé. |
Le QTc est à la fois > 500 ms et la différence par rapport à sa valeur avant traitement est >60 ms | Arrêt définitif. |
1ère apparition d’un QTc>500 ms pendant le traitement et la différence par rapport à sa valeur avant traitement est <60 ms | Interruption temporaire du traitement jusqu’à ce que le QTc repasse en dessous de 500 ms. |
2ème apparition d’un QTc>500 ms pendant le traitement et la différence par rapport à sa valeur avant traitement est <60 ms | Interruption temporaire du traitement jusqu’à ce que le QTc repasse en dessous de 500 ms. |
3ème apparition d’un QTc>500 ms pendant le traitement et la différence par rapport à sa valeur avant traitement est <60 ms | Arrêt définitif. |
Populations particulières
Population âgée
Aucune adaptation posologique particulière n’est nécessaire chez les patients âgés de plus de 65 ans.
Insuffisants rénaux
Des données limitées sont disponibles chez les patients insuffisants rénaux. Chez les patients ayant une insuffisance rénale sévère, un risque d’augmentation de l’exposition ne peut être exclu. Les patients ayant une insuffisance rénale sévère doivent être surveillés étroitement (voir rubriques 4.4 et 5.2)
Insuffisants hépatiques
Des données limitées sont disponibles chez les patients insuffisants hépatiques. Le vemurafenib étant éliminé par le foie, les patients ayant une insuffisance hépatique modérée à sévère peuvent avoir une exposition augmentée et doivent être surveillés étroitement (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité du vemurafenib n’ont pas été établies chez les enfants de moins de 18 ans. Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2, toutefois aucune recommandation posologique ne peut être établie.
Patients non caucasiens
La sécurité et l’efficacité du vemurafenib n’ont pas été établies chez des patients non caucasiens. Aucune donnée n’est disponible.
Mode d’administration
Voie orale. Les comprimés doivent être avalés entiers avec de l’eau. Ils ne doivent pas être croqués ou écrasés.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de tolérance
Les effets indésirables de tout grade les plus fréquemment rapportés (> 30 %) avec le vemurafenib ont été les suivants : arthralgies, fatigue, éruption cutanée, réaction de photosensibilité, alopécie, nausées, diarrhée, céphalées, prurit, vomissements, papillome cutané et hyperkératose. Les effets indésirables de grade 3 les plus fréquents (≥ 5%) ont été carcinome épidermoïde cutané (CEC), kératoacanthome, éruption cutanée, arthralgies et élévation de la gamma-glutamyltransférase (GGT). Les CEC ont été le plus souvent traités par exérèse locale.
Résumé tabulé des effets indésirables
Les effets indésirables rapportés chez des patients atteints d’un mélanome sont listés ci-dessous par système organe-classe MedDRA, fréquence et grade de sévérité. La convention suivante a été utilisée pour la classification des fréquences :
Très fréquent ≥ 1/10
Fréquent ≥ 1/100 à < 1/10
Peu fréquent ≥ 1/1 000 à < 1/100
Rare ≥ 1/10 000 à < 1/1 000
Très rare < 1/10 000
Les effets indésirables mentionnés dans cette rubrique sont basés sur les résultats de 468 patients issus d’une étude de phase III, randomisée, menée en ouvert chez des patients adultes présentant un mélanome non résécable ou au stade IV porteur d’une mutation BRAF V600, ainsi que d’une étude de phase II menée sur un seul groupe de patients présentant un mélanome au stade IV porteur d’une mutation BRAF V600 après échec d’au moins un traitement systémique préalable (voir rubrique 5.1). En complément, des effets indésirables issus des rapports de sécurité de tous les essais cliniques et de l’expérience depuis la commercialisation sont également rapportés. Tous les termes mentionnés sont basés sur le pourcentage le plus élevé observé lors des essais cliniques de phase II et de phase III. Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante et ont été rapportés au moyen des critères communs de toxicité NCI-CTCAE v 4.0 (common toxicity criteria) pour l’évaluation de la toxicité.
Tableau 3 : Effets indésirables survenus chez des patients traités par le vemurafenib au cours de l’étude de phase II ou de l’étude de phase III et évènements issus des rapports de sécurité de tous les essais(1) et de l’expérience depuis la commercialisation(2)
Système | Très fréquent | Fréquent | Peu fréquent | Rare |
Infection et infestations |
| Folliculite |
|
|
Tumeurs bénignes, malignes et non précisées (dont kystes et polypes) | CEC(d), kératoacanthome, kératose séborrhéique, papillome cutané | Carcinome basocellulaire, nouveau mélanome primitif(3) | Carcinome épidermoïde non cutané(1) (3) | Leucémie myélomonocy- |
Affections hématologiques et du système lymphatique |
| Neutropénie, thrombopénie(6) |
|
|
Affections de système immunitaire |
|
|
| Sarcoïdose(1)(2)(j) |
Troubles du métabolisme et de la nutrition | Diminution de l’appétit |
|
|
|
Affections du système nerveux | Céphalées, dysgueusie, vertiges | Paralysie du nerf facial, neuropathie périphérique |
|
|
Affections oculaires |
| Uvéite | Occlusion de la veine rétinienne, iridocyclite |
|
Affections vasculaires |
| Vascularite |
|
|
Affections respiratoires, thoraciques et médiastinales | Toux |
|
|
|
Affections gastro-intestinales | Diarrhée, vomissements, nausées, constipation | Stomatite | Pancréatite(2) |
|
Affections hépatobiliaires |
|
| Atteintes hépatiques(1) (2) (g) |
|
Affections de la peau et du tissu sous-cutané | Réaction de photosensibilité, kératose actinique, éruption cutanée, éruption maculopapuleuse, prurit, hyperkératose, érythème, syndrome d’érythrodysesthésie palmoplantaire, alopécie, sécheresse cutanée, érythème solaire | Eruption papuleuse, panniculite | Nécrolyse épidermique toxique(e), Syndrome de Stevens-Johnson(f) | Syndrome d'hypersensi- |
Affections musculo-squelettiques et systémiques | Arthralgie, myalgie, douleur dans les extrémités, douleur musculo-squelettique, dorsalgie | Arthrite | Maladie de Ledderhose(1)(2), maladie de Dupuytren(1)(2) |
|
Affections du rein et des voies urinaires |
|
|
| Néphrite interstitielle aiguë(1)(2) (h), nécrose tubulaire aiguë(1)(2) (h) |
Troubles généraux et anomalies au site d’administration | Fatigue, pyrexie, œdème périphérique, |
|
|
|
Investigations |
| Elévation des ALAT(c), |
|
|
Lésions, intoxications et complications liées aux procédures |
| Potentialisation de la toxicité radio-induite(1) (2) (i) |
|
|
(1) Evènements issus des rapports de sécurité de tous les essais cliniques.
(2) Evènements issus de l’expérience depuis la commercialisation.
(3) Une relation causale entre le médicament et l’évènement indésirable peut être raisonnablement possible.
(4) Progression de leucémie myélomonocytaire chronique préexistante avec mutations NRAS.
(5) Progression d’un adénocarcinome pancréatique préexistant avec mutations KRAS.
(6) Calculée sur la base des études de phase II et de phase III.
Description des effets indésirables sélectionnés
Elévation du taux d’enzymes hépatiques (c)
Les anomalies du taux d’enzymes hépatiques rapportées lors de l’étude clinique de phase III sont exprimées ci-dessous selon la proportion de patients ayant présenté une modification de grade 3 ou 4 par rapport à la valeur initiale.
Très fréquent : GGT
Fréquent : ALAT, phosphatases alcalines, bilirubine
Peu fréquent : ASAT
Aucune élévation de grade 4 des ALAT, des phosphatases alcalines ou de la bilirubine n’a été observée.
Atteintes hépatiques (g)
Sur la base de critères de détermination des atteintes hépatiques induites par un médicament élaborés par un groupe de travail international comprenant des experts cliniciens et scientifiques, les atteintes hépatiques ont été définies comme l'une des anomalies des explorations fonctionnelles suivantes:
ALAT ≥ 5x LSN
phosphatases alcalines ≥ 2x LSN (en l’absence d’autres causes d’élévation des phosphatases alcalines)
ALAT ≥ 3x LSN avec une élévation simultanée de la concentration de la bilirubine > 2x LSN
Carcinome épidermoïde cutané (d) (CEC)
Des cas de CEC ont été rapportés chez des patients traités par le vemurafenib. L’incidence des CEC chez les patients traités par le vemurafenib sur l’ensemble des études a été d’environ 20 %. La majorité des lésions retirées et examinées par un laboratoire centralisé indépendant d’anatomopathologie a été classée comme CE de sous-type kératoacanthome ou présentant des caractéristiques de kératoacanthome mixte (52 %). La plupart des lésions classées comme « autres » (43 %) étaient des lésions cutanées bénignes (par exemple verrue banale, kératose actinique, kératose bénigne, kyste/kyste bénin). Les CEC sont habituellement apparus en début de traitement avec un délai médian de première apparition de 7 à 8 semaines. Parmi les patients chez lesquels un CEC est apparu, environ 33 % ont présenté plus d’une apparition avec un délai médian de 6 semaines entre les apparitions. Les cas de CEC ont été le plus souvent pris en charge par une simple exérèse, et les patients ont généralement continué le traitement sans modification de la dose (voir rubriques 4.2 et 4.4).
Carcinome épidermoïde non cutané
Des cas de carcinome épidermoïde non cutané ont été rapportés chez des patients ayant reçu du vemurafenib dans le cadre des essais cliniques. Une surveillance visant à détecter l’apparition de carcinome épidermoïde non cutané doit être réalisée comme décrit à la rubrique 4.4.
Nouveau mélanome primitif
Des nouveaux mélanomes primitifs ont été rapportés dans les essais cliniques. Ces cas ont été pris en charge par exérèse et les patients ont poursuivis leur traitement sans adaptation posologique. Une surveillance visant à détecter l’apparition de lésions cutanées doit être effectuée comme indiqué à la rubrique 4.4.
Potentialisation de la toxicité radio-induite(i)
Les cas rapportés comprennent phénomène de rappel, lésion cutanée radique, pneumopathie radique, oesophagite radique, proctite radique, hépatite radique, cystite radique et radionécrose.
Dans un essai clinique de phase III (MO25515, N = 3219), une incidence plus importante de potentialisation de la toxicité radio-induite a été rapportée chez les patients ayant reçu une radiothérapie avant ou pendant le traitement par vemurafenib (9,1%) par rapport aux patients ayant reçu une radiothérapie et le vemurafenib de manière concomitante (5,2%) ou chez ceux dont la radiothérapie était antérieure au vemurafenib (1,5%).
Réactions d’hypersensibilité(e)
Des réactions graves d’hypersensibilité, dont des cas d’anaphylaxie, ont été rapportées lors d’un traitement par le vemurafenib. Les réactions sévères d’hypersensibilité peuvent comprendre un syndrome de Stevens-Johnson, une éruption cutanée généralisée, un érythème ou une hypotension. Le traitement par le vemurafenib doit être définitivement arrêté chez tout patient présentant une réaction sévère d’hypersensibilité (voir rubrique 4.4).
Réactions cutanée sévères (f)
Des réactions cutanées sévères ont été rapportées chez les patients recevant du vemurafenib, dont des cas rares de syndrome de Stevens-Johnson et de nécrolyse épidermique toxique dans l’essai clinique pivot. Le traitement par le vemurafenib doit être définitivement arrêté chez tout patient présentant une réaction cutanée sévère.
Allongement de l’intervalle QT
Une analyse des données ECG centralisées issues d’une sous-étude de phase II non contrôlée, en ouvert, sur l’intervalle QT menée chez 132 patients traités par 960 mg de vemurafenib deux fois par jour (NP22657) a montré un allongement de l’intervalle QTc exposition-dépendante. L’effet moyen sur l’intervalle QTc est demeuré stable entre 12-15 ms au-delà du premier mois de traitement ; le plus important allongement moyen de QTc (15,1 ms ; limite supérieure de l’IC à 95 % : 17,7 ms) a été observée au cours des 6 premiers mois (n = 90 patients). Deux patients (1,5 %) ont présenté des valeurs absolues de QTc > 500 ms (grade 3 CTC) en cours de traitement, et seul un patient (0,8 %) a présenté une modification de QTc > 60 ms par rapport à la valeur initiale (voir rubrique 4.4).
Atteintes rénales aiguës (h)
Des cas de toxicité rénale ont été rapportés avec le vemurafenib allant d’élévations de la créatinine sérique à une néphrite interstitielle aiguë et une nécrose tubulaire aiguë, certains observés dans un contexte de déshydratation. Les élévations de la créatinine sérique étaient généralement d'intensité légère (> 1 à 1,5x LSN) à modérée (>1,5 à 3 x LSN) et étaient de nature réversible (voir tableau 4).
Tableau 4: Changements de la créatinine par rapport à la valeur basale dans l’essai de phase III
| Vemurafenib (%) | Dacarbazine (%) |
Changement de grade 1 par rapport à la valeur basale jusqu’à tous grades | 27,9 | 6,1 |
Changement de grade 1 par rapport à la valeur basale jusqu’à un grade 3 ou plus | 1,2 | 1,1 |
jusqu’à un grade 3 | 0,3 | 0,4 |
jusqu’à un grade 4 | 0,9 | 0,8 |
Tableau 5: Cas d’atteintes rénales aiguës dans l’essai de phase III
| Vemurafenib (%) | Dacarbazine (%) |
Cas d’atteintes rénales aiguës* | 10,0 | 1,4 |
Cas d’atteintes rénales aiguës associés à des évènements de déshydratation | 5,5 | 1,0 |
Adaptation posologique suite à une atteinte rénale aiguë | 2,1 | 0 |
Tous les pourcentages sont exprimés en nombre de cas sur l'ensemble des patients exposés à chaque médicament.
* y compris atteinte rénale aiguë, insuffisance rénale, et anomalies du bilan biologique évoquant une atteinte rénale aiguë.
Sarcoïdose(j)
Des cas de sarcoïdose ont été rapportés chez des patients traités par le vemurafenib, impliquant principalement la peau, les poumons et les yeux. Dans la majorité des cas, le traitement par le vemurafenib a été maintenu et l'événement de sarcoïdose soit s’est résolu soit a persisté.
Populations particulières
Population âgée
Dans l’étude de phase III, quatre-vingt-quatorze (28 %) des 336 patients atteints d’un mélanome non résécable ou métastatique traités par le vemurafenib étaient âgés de ≥ 65 ans. La probabilité de survenue d’effets indésirables, dont CEC, diminution de l’appétit et troubles cardiaques, peut être plus élevée chez les patients âgés (≥ 65 ans).
Sexe
Durant les essais cliniques menés avec le vemurafenib, les effets indésirables de grade 3 plus fréquemment rapportés chez les femmes que chez les hommes étaient éruption cutanée, arthralgie et photosensibilité.
Population pédiatrique
La sécurité du vemurafenib n’a pas été établie chez les enfants et les adolescents. Dans un essai clinique mené chez 6 patients adolescents, aucun nouveau signal de sécurité n’a été observé.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHE
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
EU/1/12/751/001
10. DATE DE MISE À JOUR DU TEXTE
18 mars 2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu/
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 2910354 | ZELBORAF 240 MG COMP PELL 56 X 240 MG UD | L01EC01 | - | € 1353,13 | Oui | - | - |